Vibrationally resolved electronic spectra with ADF¶
In this tutorial we use the vertical gradient Franck-Condon (VG-FC) Vibronic-Structure Tracking (VST) method to calculate the vibrationally resolved absorption spectrum of the first excited singlet state of naphthalene.
There are different methods to calculate a vibrationally resolved absorption spectrum. Out of these methods VST is in principal the quickest method and can also be used for much larger sized molecules. It is based on a mode-tracking algorithm and works by tracking those modes that are expected to have the largest impact on the vibronic-structure of the spectrum. More information on VST and related methods can be found in the AMS user manual:
Step 1: Geometry Optimization¶
Let us first obtain a naphthalene molecule, and optimize its geometry with ADF.
 and type “naphthalene” into the box.
and type “naphthalene” into the box. to check the symmetry (should be D(2H))
to check the symmetry (should be D(2H))
Step 2: Excited state gradient¶
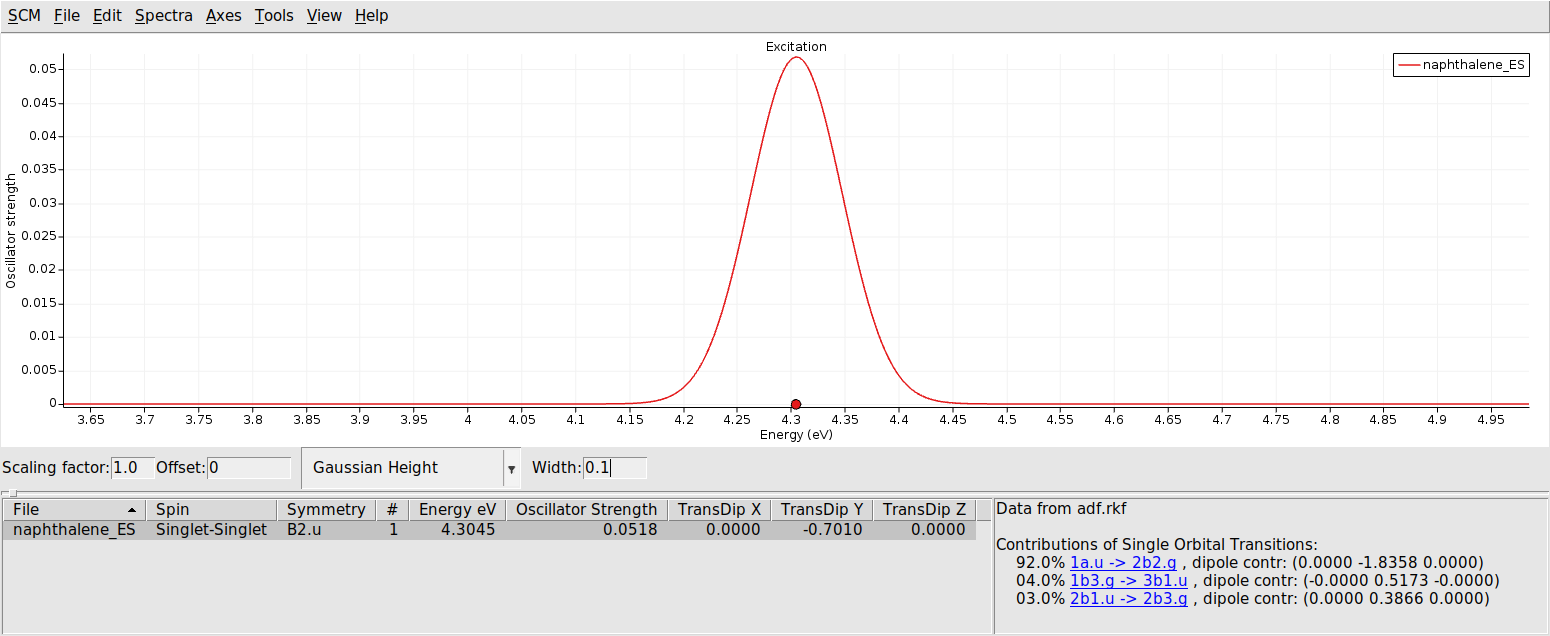
Here we will look at the vibrationally resolved absorption spectra of the lowest electronically excited singlet state S1. The VG-FC Vibronic-Structure Tracking method needs the excited state gradient of S1 at the ground state geometry.
See also
ADF manual sections on excitations and excited states gradients
Step 3: Vibronic-Structure Tracking¶
The calculated lowest excited singlet state is of B2.u symmetry. For the VG-FC vibronic-structure tracking method we need a new input:
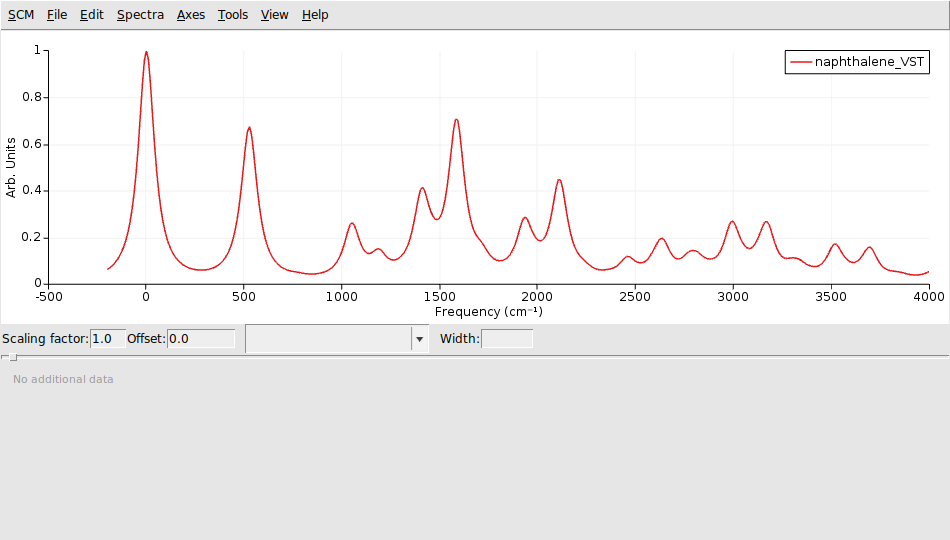
The spectrum is relative to the 0-0 excitation energy. The default (artificial) broadening is relatively wide, therefore it was changed to 50 cm-1.