Volume Scan with Birch-Murnaghan Equation of State¶
Run a lattice PES scan for diamond with ReaxFF, fit the energy-volume curve to a Birch-Murnaghan equation of state, and rescale the cell to the fitted equilibrium volume.
Initial structure¶
To get the initial diamond structure in AMSinput:
Builders -> Crystal Structure -> Cubic -> Diamond
Click “Ok”
File -> “Export Coordinates (System)” -> “.in…”
Copy the contents of the resulting .in file
from scm.base import ChemicalSystem
from scm.plams import view
diamond = ChemicalSystem("""
System
Atoms
C -0.44625 -0.44625 -0.44625
C 0.44625 0.44625 0.44625
End
Lattice
0.0 1.785 1.785
1.785 0.0 1.785
1.785 1.785 0.0
End
End
""")
view(diamond, show_lattice_vectors=True, direction="tilt_x", guess_bonds=True)
Run a volume scan for the diamond structure¶
from scm.plams import Settings, AMSJob
sett = Settings()
sett.input.ReaxFF.ForceField = "C.ff"
sett.input.ams.Task = "PESScan"
sett.input.ams.PESScan.ScanCoordinate.CellVolumeScalingRange = "0.8 1.3"
sett.input.ams.PESScan.ScanCoordinate.nPoints = 11
job = AMSJob(name="DiamondVolumeScan", settings=sett, molecule=diamond)
job.run();
[22.06|14:05:56] JOB DiamondVolumeScan STARTED
[22.06|14:05:56] JOB DiamondVolumeScan RUNNING
[22.06|14:05:57] JOB DiamondVolumeScan FINISHED
[22.06|14:05:57] JOB DiamondVolumeScan SUCCESSFUL
Extract the results¶
Obtain the volumes in Å3
Obtain the energies in eV
Plot the results and fit with the Birch-Murnaghan EoS
from scm.utils.conversions import plams_molecule_to_chemsys
from scm.base import Units
from ase.eos import EquationOfState
import matplotlib.pyplot as plt
res = job.results.get_pesscan_results(molecules=True)
# Obtain volumes
volumes = [plams_molecule_to_chemsys(x).lattice.get_volume() for x in res["Molecules"]]
# Obtain energies
energy_unit_conversion = Units.conversion_factor("Ha", "eV")
energies = [energy * energy_unit_conversion for energy in res["PES"]]
# Fit and plot
eos = EquationOfState(volumes, energies, eos="birchmurnaghan")
v0, e0, B = eos.fit()
eos.plot(show=True);
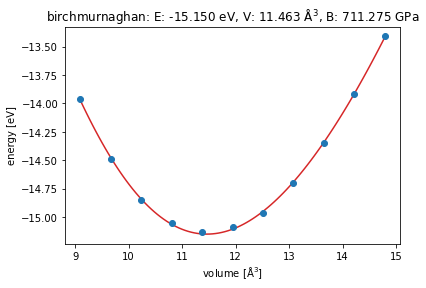
print(f"{e0=:8.3f} eV\n{v0=:8.3f} Å\u00b3\n{ B=:8.3f} GPa")
e0= -15.150 eV
v0= 11.463 ų
B= 4.439 GPa
Scale cell to match minimum volume¶
set_density applies uniform scaling.
diamond_scaled = diamond.copy()
diamond_scaled.set_density(diamond.total_mass() / v0)
print(diamond_scaled)
System
Atoms
C -0.4473943191070795 -0.4473943191070795 -0.4473943191070795
C 0.4473943191070795 0.4473943191070795 0.4473943191070795
End
... output trimmed ....
0 1.7895772764283184 1.7895772764283184
1.7895772764283184 0 1.7895772764283184
1.7895772764283184 1.7895772764283184 0
End
End
print(f"{diamond.lattice.get_volume()=:.3f}")
print(f"{diamond_scaled.lattice.get_volume()=:.3f}")
diamond.lattice.get_volume()=11.375
diamond_scaled.lattice.get_volume()=11.463
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial structure
# To get the initial diamond structure in AMSinput:
#
# 1. Builders -> Crystal Structure -> Cubic -> Diamond
# 2. Click "Ok"
# 3. File -> "Export Coordinates (System)" -> ".in..."
# 4. Copy the contents of the resulting .in file
from scm.base import ChemicalSystem
from scm.plams import view
diamond = ChemicalSystem("""
System
Atoms
C -0.44625 -0.44625 -0.44625
C 0.44625 0.44625 0.44625
End
Lattice
0.0 1.785 1.785
1.785 0.0 1.785
1.785 1.785 0.0
End
End
""")
view(diamond, show_lattice_vectors=True, direction="tilt_x", guess_bonds=True, picture_path="picture1.png")
# ## Run a volume scan for the diamond structure
from scm.plams import Settings, AMSJob
sett = Settings()
sett.input.ReaxFF.ForceField = "C.ff"
sett.input.ams.Task = "PESScan"
sett.input.ams.PESScan.ScanCoordinate.CellVolumeScalingRange = "0.8 1.3"
sett.input.ams.PESScan.ScanCoordinate.nPoints = 11
job = AMSJob(name="DiamondVolumeScan", settings=sett, molecule=diamond)
job.run()
# ## Extract the results
#
# - Obtain the volumes in Å<sup>3</sup>
# - Obtain the energies in eV
# - Plot the results and fit with the Birch-Murnaghan EoS
from scm.utils.conversions import plams_molecule_to_chemsys
from scm.base import Units
from ase.eos import EquationOfState
import matplotlib.pyplot as plt
res = job.results.get_pesscan_results(molecules=True)
# Obtain volumes
volumes = [plams_molecule_to_chemsys(x).lattice.get_volume() for x in res["Molecules"]]
# Obtain energies
energy_unit_conversion = Units.conversion_factor("Ha", "eV")
energies = [energy * energy_unit_conversion for energy in res["PES"]]
# Fit and plot
eos = EquationOfState(volumes, energies, eos="birchmurnaghan")
v0, e0, B = eos.fit()
eos.plot(show=True)
print(f"{e0=:8.3f} eV\n{v0=:8.3f} Å\u00b3\n{ B=:8.3f} GPa")
# ## Scale cell to match minimum volume
#
# `set_density` applies uniform scaling.
diamond_scaled = diamond.copy()
diamond_scaled.set_density(diamond.total_mass() / v0)
print(diamond_scaled)
print(f"{diamond.lattice.get_volume()=:.3f}")
print(f"{diamond_scaled.lattice.get_volume()=:.3f}")