Band structure with BAND (HSE06, DFT+U)¶
Band structure with the hybrid HSE06 functional with BAND DFT¶
Hybrid functionals often give a better band gap than semilocal functionals.
For band structures always use the primitive unit cell. Here we build the primitive unit cell of diamond:
Primitive unit cell of diamond¶
from scm.base import ChemicalSystem
from scm.plams import view
d = 1.785
cs = ChemicalSystem(
f"""
System
Atoms
C 0 0 0
C {d/2} {d/2} {d/2}
End
Lattice
0.0 {d} {d}
{d} 0.0 {d}
{d} {d} 0.0
End
End"""
).make_primitive_cell()
# here make_primitive_cell() is unnecessary because it is already the primitive cell
# note that make_primitive_cell() may change the coordinates and lattice vectors even if the cell is already primitive
cs.guess_bonds() # does not affect BAND DFT calculation, just for visualization
view(cs, width=150, height=150) # visualize in Jupyter notebook
HSE06 settings for the BAND engine¶
Set up the DFT settings for HSE06 with BAND and calculate the band structure:
from scm.plams import Settings, AMSJob
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.band.XC.libxc = "hse06" # hse06 functional from libxc
s.input.band.Basis.Core = "None" # no frozen core
s.input.band.Basis.Type = "DZ" # very small basis set, make larger for production
s.input.band.NumericalQuality = "Basic" # improve for production
s.input.band.KSPace.Regular.NumberOfPoints = "3 3 3" # improve for production
s.input.band.BandStructure.Enabled = "Yes"
job = AMSJob(settings=s, molecule=cs, name="diamond-hse06")
job.run();
[09.02|16:26:55] JOB diamond-hse06 STARTED
[09.02|16:26:55] JOB diamond-hse06 RUNNING
[09.02|16:31:10] JOB diamond-hse06 FINISHED
[09.02|16:31:10] JOB diamond-hse06 SUCCESSFUL
Get band structure data from results and plot them¶
from scm.plams.tools.plot import plot_band_structure
x, y_spin_up, y_spin_down, labels, fermi_energy = job.results.get_band_structure(unit="eV")
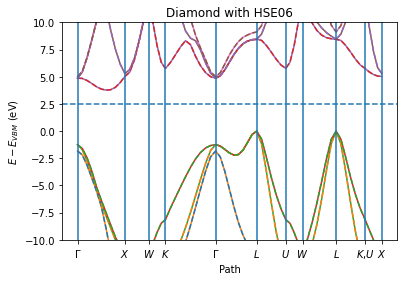
ax = plot_band_structure(x, y_spin_up, y_spin_down, labels, fermi_energy, zero="vbmax")
ax.set_ylim(-10, 10)
ax.set_ylabel("$E - E_{VBM}$ (eV)")
ax.set_xlabel("Path")
ax.set_title("Diamond with HSE06")
ax;
Extract the VBM and CBM. Important note: These values are taken from the self-consistent field calculation and the k-points used there, not the band structure calculation!
from scm.base import Units
ha2ev = Units.conversion_factor("hartree", "eV")
try:
vbm = ha2ev * job.results.readrkf("BandStructure", "TopValenceBand", file="engine")
cbm = ha2ev * job.results.readrkf("BandStructure", "BottomConductionBand", file="engine")
gap = cbm - vbm
print(f"Valence band maximum: {vbm=:.2f} eV")
print(f"Conduction band minimum: {cbm=:.2f} eV")
print(f"Band gap: {gap=:.2f} eV")
except KeyError:
print("Couldn't extract VBM and CBM")
Valence band maximum: vbm=-5.18 eV
Conduction band minimum: cbm=-0.18 eV
Band gap: gap=5.01 eV
Spin-polarized (unrestricted) band structures with BAND DFT¶
If you perform a spin-polarized calculation you get both spin-up and spin-down bands. Below a spin-polarized DFT+U calculation on NiO is performed together with the BAND DFT engine.
For band structures always use the primitive unit cell. Here we build the primitive unit cell of NiO.
Primitive unit cell of NiO¶
from scm.base import ChemicalSystem
from scm.plams import view
d = 2.085
cs = ChemicalSystem(
f"""
System
Atoms
Ni 0.0 0.0 0.0
O {d} {d} {d}
End
Lattice
0.0 {d} {d}
{d} 0.0 {d}
{d} {d} 0.0
End
End"""
).make_primitive_cell()
cs.guess_bonds() # does not affect BAND DFT calculation, just for visualization
view(cs, direction="along_z", width=150, height=150, show_atom_labels=True)

DFT+U settings with spin polarization (unrestricted)¶
Next, set up the DFT+U settings for a SinglePoint calculation with band structure:
from scm.plams import Settings, AMSJob
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.band.Unrestricted = "Yes"
s.input.band.XC.GGA = "BP86"
s.input.band.Basis.Type = "DZ"
s.input.band.NumericalQuality = "Basic"
s.input.band.HubbardU.Atom = [
Settings(Element="Ni", UValue=0.6, LValue="d"), # UValue in hartree
# Settings(....) for other elements if needed
]
s.input.band.BandStructure.Enabled = "Yes"
job = AMSJob(settings=s, molecule=cs, name="NiO")
job.run();
[09.02|16:34:20] JOB NiO STARTED
[09.02|16:34:20] JOB NiO RUNNING
[09.02|16:34:58] JOB NiO FINISHED
[09.02|16:34:58] JOB NiO SUCCESSFUL
Get the band structure data and plot¶
from scm.plams.tools.plot import plot_band_structure
x, y_spin_up, y_spin_down, labels, fermi_energy = job.results.get_band_structure(unit="eV")
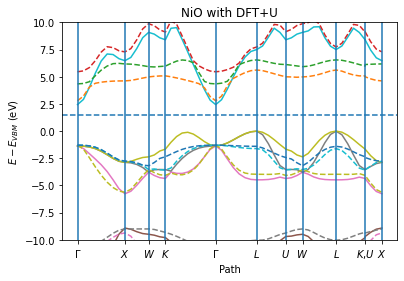
ax = plot_band_structure(x, y_spin_up, y_spin_down, labels, fermi_energy, zero="vbmax")
ax.set_ylim(-10, 10)
ax.set_ylabel("$E - E_{VBM}$ (eV)")
ax.set_xlabel("Path")
ax.set_title("NiO with DFT+U")
ax;
Extract the VBM and CBM. Important note: These values are taken from the self-consistent field calculation and the k-points used there, not the band structure calculation!
from scm.base import Units
ha2ev = Units.conversion_factor("hartree", "eV")
try:
vbm = ha2ev * job.results.readrkf("BandStructure", "TopValenceBand", file="engine")
cbm = ha2ev * job.results.readrkf("BandStructure", "BottomConductionBand", file="engine")
gap = cbm - vbm
print(f"Valence band maximum: {vbm=:.2f} eV")
print(f"Conduction band minimum: {cbm=:.2f} eV")
print(f"Band gap: {gap=:.2f} eV")
except KeyError:
print("Couldn't extract VBM and CBM")
Valence band maximum: vbm=-6.31 eV
Conduction band minimum: cbm=-4.36 eV
Band gap: gap=1.95 eV
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Band structure with the hybrid HSE06 functional with BAND DFT
#
# Hybrid functionals often give a better band gap than semilocal functionals.
#
# For band structures **always** use the **primitive unit cell**. Here we build the primitive unit cell of diamond:
# ### Primitive unit cell of diamond
from scm.base import ChemicalSystem
from scm.plams import view
d = 1.785
cs = ChemicalSystem(
f"""
System
Atoms
C 0 0 0
C {d/2} {d/2} {d/2}
End
Lattice
0.0 {d} {d}
{d} 0.0 {d}
{d} {d} 0.0
End
End"""
).make_primitive_cell()
# here make_primitive_cell() is unnecessary because it is already the primitive cell
# note that make_primitive_cell() may change the coordinates and lattice vectors even if the cell is already primitive
cs.guess_bonds() # does not affect BAND DFT calculation, just for visualization
view(cs, width=150, height=150, picture_path="picture1.png") # visualize in Jupyter notebook
# ### HSE06 settings for the BAND engine
# Set up the DFT settings for HSE06 with BAND and calculate the band structure:
from scm.plams import Settings, AMSJob
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.band.XC.libxc = "hse06" # hse06 functional from libxc
s.input.band.Basis.Core = "None" # no frozen core
s.input.band.Basis.Type = "DZ" # very small basis set, make larger for production
s.input.band.NumericalQuality = "Basic" # improve for production
s.input.band.KSPace.Regular.NumberOfPoints = "3 3 3" # improve for production
s.input.band.BandStructure.Enabled = "Yes"
job = AMSJob(settings=s, molecule=cs, name="diamond-hse06")
job.run()
# ### Get band structure data from results and plot them
from scm.plams.tools.plot import plot_band_structure
x, y_spin_up, y_spin_down, labels, fermi_energy = job.results.get_band_structure(unit="eV")
ax = plot_band_structure(x, y_spin_up, y_spin_down, labels, fermi_energy, zero="vbmax")
ax.set_ylim(-10, 10)
ax.set_ylabel("$E - E_{VBM}$ (eV)")
ax.set_xlabel("Path")
ax.set_title("Diamond with HSE06")
ax
ax.figure.savefig("picture2.png")
# Extract the VBM and CBM. Important note: These values are taken from the self-consistent field calculation and the k-points used there, not the band structure calculation!
from scm.base import Units
ha2ev = Units.conversion_factor("hartree", "eV")
try:
vbm = ha2ev * job.results.readrkf("BandStructure", "TopValenceBand", file="engine")
cbm = ha2ev * job.results.readrkf("BandStructure", "BottomConductionBand", file="engine")
gap = cbm - vbm
print(f"Valence band maximum: {vbm=:.2f} eV")
print(f"Conduction band minimum: {cbm=:.2f} eV")
print(f"Band gap: {gap=:.2f} eV")
except KeyError:
print("Couldn't extract VBM and CBM")
# ## Spin-polarized (unrestricted) band structures with BAND DFT
#
# If you perform a spin-polarized calculation you get both spin-up and spin-down bands. Below a spin-polarized DFT+U calculation on NiO is performed together with the BAND DFT engine.
#
# For band structures **always** use the **primitive unit cell**. Here we build the primitive unit cell of NiO.
#
# ### Primitive unit cell of NiO
from scm.base import ChemicalSystem
from scm.plams import view
d = 2.085
cs = ChemicalSystem(
f"""
System
Atoms
Ni 0.0 0.0 0.0
O {d} {d} {d}
End
Lattice
0.0 {d} {d}
{d} 0.0 {d}
{d} {d} 0.0
End
End"""
).make_primitive_cell()
cs.guess_bonds() # does not affect BAND DFT calculation, just for visualization
view(cs, direction="along_z", width=150, height=150, show_atom_labels=True, picture_path="picture3.png")
# ### DFT+U settings with spin polarization (unrestricted)
# Next, set up the DFT+U settings for a SinglePoint calculation with band structure:
from scm.plams import Settings, AMSJob
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.band.Unrestricted = "Yes"
s.input.band.XC.GGA = "BP86"
s.input.band.Basis.Type = "DZ"
s.input.band.NumericalQuality = "Basic"
s.input.band.HubbardU.Atom = [
Settings(Element="Ni", UValue=0.6, LValue="d"), # UValue in hartree
# Settings(....) for other elements if needed
]
s.input.band.BandStructure.Enabled = "Yes"
job = AMSJob(settings=s, molecule=cs, name="NiO")
job.run()
# ### Get the band structure data and plot
from scm.plams.tools.plot import plot_band_structure
x, y_spin_up, y_spin_down, labels, fermi_energy = job.results.get_band_structure(unit="eV")
ax = plot_band_structure(x, y_spin_up, y_spin_down, labels, fermi_energy, zero="vbmax")
ax.set_ylim(-10, 10)
ax.set_ylabel("$E - E_{VBM}$ (eV)")
ax.set_xlabel("Path")
ax.set_title("NiO with DFT+U")
ax
ax.figure.savefig("picture4.png")
# Extract the VBM and CBM. Important note: These values are taken from the self-consistent field calculation and the k-points used there, not the band structure calculation!
from scm.base import Units
ha2ev = Units.conversion_factor("hartree", "eV")
try:
vbm = ha2ev * job.results.readrkf("BandStructure", "TopValenceBand", file="engine")
cbm = ha2ev * job.results.readrkf("BandStructure", "BottomConductionBand", file="engine")
gap = cbm - vbm
print(f"Valence band maximum: {vbm=:.2f} eV")
print(f"Conduction band minimum: {cbm=:.2f} eV")
print(f"Band gap: {gap=:.2f} eV")
except KeyError:
print("Couldn't extract VBM and CBM")