Band structure with Quantum ESPRESSO (PBE)¶
Band structure for ferromagnetic iron with Quantum ESPRESSO¶
For band structures, always use the primitive unit cell.
Primitive unit cell of Fe¶
from scm.base import ChemicalSystem
from scm.plams import view
d = 1.435
cs = ChemicalSystem(
f"""
System
Atoms
Fe 0.0 0.0 0.0
End
Lattice
{-d} {d} {d}
{d} {-d} {d}
{d} {d} {-d}
End
End"""
)
cs.guess_bonds() # does not affect the Quantum ESPRESSO DFT engine, just for visualization
view(cs, width=150, height=150)
Set up PBE band structure calculation with Quantum ESPRESSO¶
from scm.plams import Settings, AMSJob
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.QuantumESPRESSO = Settings()
s.input.QuantumESPRESSO.BandStructure.K_Points._h = "ams_kpath"
s.input.QuantumESPRESSO.BandStructure.K_Points._1 = ""
s.input.QuantumESPRESSO.DOS.PDOS = "True"
s.input.QuantumESPRESSO.K_Points._h = "automatic"
s.input.QuantumESPRESSO.K_Points._1 = "8 8 8 0 0 0"
s.input.QuantumESPRESSO.Properties.BandStructure = "True"
s.input.QuantumESPRESSO.Properties.DOS = "True"
s.input.QuantumESPRESSO.Pseudopotentials.Family = "pslibrary-US"
s.input.QuantumESPRESSO.Pseudopotentials.Functional = "PBE"
s.input.QuantumESPRESSO.System.degauss = "0.01"
s.input.QuantumESPRESSO.System.ecutwfc = "50.0"
s.input.QuantumESPRESSO.System.nspin = "Collinear"
s.input.QuantumESPRESSO.System.occupations = "Smearing"
s.input.QuantumESPRESSO.System.starting_magnetization = [Settings()]
s.input.QuantumESPRESSO.System.starting_magnetization[0].Label = "Fe"
s.input.QuantumESPRESSO.System.starting_magnetization[0].Value = "1.0"
s.runscript.preamble_lines = [
"export SCM_DISABLE_MPI=1"
] # needed for Quantum ESPRESSO calculations with AMS
s.runscript.postamble_lines = []
job = AMSJob(settings=s, molecule=cs, name="ferromagnetic-Fe")
job.run();
[09.02|16:58:07] JOB ferromagnetic-Fe STARTED
[09.02|16:58:07] JOB ferromagnetic-Fe RUNNING
[09.02|16:58:20] JOB ferromagnetic-Fe FINISHED
[09.02|16:58:20] JOB ferromagnetic-Fe SUCCESSFUL
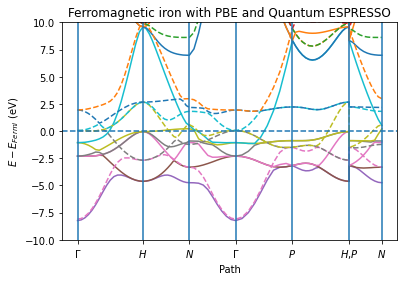
Get the band structure data and plot it¶
from scm.plams.tools.plot import plot_band_structure
x, y_spin_up, y_spin_down, labels, fermi_energy = job.results.get_band_structure(unit="eV")
ax = plot_band_structure(x, y_spin_up, y_spin_down, labels, fermi_energy, zero="fermi")
ax.set_ylim(-10, 10)
ax.set_ylabel("$E - E_{Fermi}$ (eV)")
ax.set_xlabel("Path")
ax.set_title("Ferromagnetic iron with PBE and Quantum ESPRESSO")
ax;
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Band structure for ferromagnetic iron with Quantum ESPRESSO
#
# For band structures, **always** use the **primitive unit cell**.
#
# ### Primitive unit cell of Fe
from scm.base import ChemicalSystem
from scm.plams import view
d = 1.435
cs = ChemicalSystem(
f"""
System
Atoms
Fe 0.0 0.0 0.0
End
Lattice
{-d} {d} {d}
{d} {-d} {d}
{d} {d} {-d}
End
End"""
)
cs.guess_bonds() # does not affect the Quantum ESPRESSO DFT engine, just for visualization
view(cs, width=150, height=150, picture_path="picture1.png")
# ### Set up PBE band structure calculation with Quantum ESPRESSO
from scm.plams import Settings, AMSJob
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.QuantumESPRESSO = Settings()
s.input.QuantumESPRESSO.BandStructure.K_Points._h = "ams_kpath"
s.input.QuantumESPRESSO.BandStructure.K_Points._1 = ""
s.input.QuantumESPRESSO.DOS.PDOS = "True"
s.input.QuantumESPRESSO.K_Points._h = "automatic"
s.input.QuantumESPRESSO.K_Points._1 = "8 8 8 0 0 0"
s.input.QuantumESPRESSO.Properties.BandStructure = "True"
s.input.QuantumESPRESSO.Properties.DOS = "True"
s.input.QuantumESPRESSO.Pseudopotentials.Family = "pslibrary-US"
s.input.QuantumESPRESSO.Pseudopotentials.Functional = "PBE"
s.input.QuantumESPRESSO.System.degauss = "0.01"
s.input.QuantumESPRESSO.System.ecutwfc = "50.0"
s.input.QuantumESPRESSO.System.nspin = "Collinear"
s.input.QuantumESPRESSO.System.occupations = "Smearing"
s.input.QuantumESPRESSO.System.starting_magnetization = [Settings()]
s.input.QuantumESPRESSO.System.starting_magnetization[0].Label = "Fe"
s.input.QuantumESPRESSO.System.starting_magnetization[0].Value = "1.0"
s.runscript.preamble_lines = ["export SCM_DISABLE_MPI=1"] # needed for Quantum ESPRESSO calculations with AMS
s.runscript.postamble_lines = []
job = AMSJob(settings=s, molecule=cs, name="ferromagnetic-Fe")
job.run()
# ### Get the band structure data and plot it
from scm.plams.tools.plot import plot_band_structure
x, y_spin_up, y_spin_down, labels, fermi_energy = job.results.get_band_structure(unit="eV")
ax = plot_band_structure(x, y_spin_up, y_spin_down, labels, fermi_energy, zero="fermi")
ax.set_ylim(-10, 10)
ax.set_ylabel("$E - E_{Fermi}$ (eV)")
ax.set_xlabel("Path")
ax.set_title("Ferromagnetic iron with PBE and Quantum ESPRESSO")
ax
ax.figure.savefig("picture2.png")