M3GNet Universal Potential: M3GNet-UP-2022¶
Run a geometry optimization with the default M3GNet-UP-2022 model in AMS via PLAMS.
Downloads: Notebook | Script ?
Requires: AMS2026 or later
Required AMS packages: m3gnet ?
GUI tutorial: M3GNet cohesive energy
Related Documentation
Related Examples

M3GNet-UP-2022 cohesive energy (naphthalene)¶
This PLAMS workflow mirrors the GUI tutorial and uses the same structures, settings, and steps:
Gas-phase naphthalene geometry optimization
Crystal naphthalene lattice + geometry optimization
D3(BJ) dispersion add-on
Cohesive energy calculation from the two energies
1) Gas-phase naphthalene structure¶
from scm.base import ChemicalSystem
from scm.plams import view
naph_gas = ChemicalSystem(
"""
System
Atoms
C -2.43705000 0.70853000 0.00000000
C -1.24580000 1.40435000 0.00000000
C -1.24580000 -1.40435000 0.00000000
C 0.00000000 0.71830000 0.00000000
C -2.43705000 -0.70853000 0.00000000
C 0.00000000 -0.71830000 0.00000000
C 1.24580000 1.40435000 0.00000000
C 2.43705000 0.70853000 0.00000000
C 2.43705000 -0.70853000 0.00000000
C 1.24580000 -1.40435000 0.00000000
H -1.24407000 2.49615000 0.00000000
H -3.38504000 1.24787000 0.00000000
H -3.38504000 -1.24787000 0.00000000
H -1.24407000 -2.49615000 0.00000000
H 1.24407000 -2.49615000 0.00000000
H 3.38504000 -1.24787000 0.00000000
H 3.38504000 1.24787000 0.00000000
H 1.24407000 2.49615000 0.00000000
End
End
"""
)
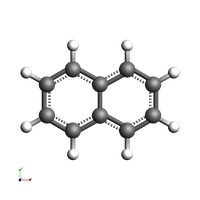
view(naph_gas, direction="along_pca3", guess_bonds=True, width=200, height=200)
3) Gas-phase naphthalene (geometry optimization)¶
Matches the GUI steps for the gas-phase molecule.
from scm.plams import AMSJob
s_gas = m3gnet_settings.copy()
s_gas.input.ams.Task = "GeometryOptimization"
job_gas = AMSJob(settings=s_gas, molecule=naph_gas, name="naphthalene_gas")
job_gas.run();
[11.02|11:35:06] JOB naphthalene_gas STARTED
[11.02|11:35:06] JOB naphthalene_gas RUNNING
[11.02|11:35:14] JOB naphthalene_gas FINISHED
[11.02|11:35:15] JOB naphthalene_gas SUCCESSFUL
4) Crystalline naphthalene (lattice + geometry optimization)¶
Matches the GUI steps for the crystal, including Optimize lattice → Yes.
naph_crystal = ChemicalSystem(
"""
System
Atoms
C -0.87388368 0.11073012 2.36589887
C 0.15273024 3.05568012 4.76889870
C 4.17013024 5.77916988 4.76889870
C 3.14351632 2.83421988 2.36589887
H -1.09410897 0.35103804 3.23705766
H 0.37295554 3.29598804 3.89773991
H 4.39035554 5.53886196 3.89773991
H 2.92329103 2.59391196 3.23705766
C -0.12579372 0.96653259 1.59034638
C -0.59535971 3.91148259 5.54445119
C 3.42204029 4.92336741 5.54445119
C 3.89160628 1.97841741 1.59034638
H 0.15997330 1.77874980 1.94280538
H -0.88112673 4.72369980 5.19199219
H 3.13627327 4.11115020 5.19199219
H 4.17737330 1.16620020 1.94280538
C 0.21357858 0.61726152 0.25185835
C -0.93473202 3.56221152 6.88293921
C 3.08266798 5.27263848 6.88293921
C 4.23097858 2.32768848 0.25185835
C -3.75675609 1.48307682 6.55901941
C 3.03560266 4.42802682 0.57577816
C 7.05300266 4.40682318 0.57577816
C 0.26064391 1.46187318 6.55901941
H -3.47488266 2.30353989 6.89435489
H 2.75372923 5.24848989 0.24044268
H 6.77112923 3.58636011 0.24044268
H 0.54251734 0.64141011 6.89435489
C 6.72508585 4.75845021 1.84862605
C 0.58856072 1.81350021 5.28617152
C -3.42883928 1.13144979 5.28617152
C 2.70768585 4.07639979 1.84862605
H 6.21662119 4.18477395 2.37588759
H 1.09702538 1.23982395 4.75890998
H -2.92037462 1.70512605 4.75890998
H 2.19922119 4.65007605 2.37588759
End
Lattice
8.03480000 0.00000000 0.00000000
0.00000000 5.88990000 0.00000000
-4.73855343 0.00000000 7.13479757
End
End
"""
)
naph_crystal_supercell = naph_crystal.make_supercell((2, 2, 2)) # for visualization
view(naph_crystal_supercell, direction="tilt_y", guess_bonds=True, width=200, height=200)
s_crystal = m3gnet_settings.copy()
# Geometry optimization with lattice optimization enabled
s_crystal.input.ams.Task = "GeometryOptimization"
s_crystal.input.ams.GeometryOptimization.OptimizeLattice = "Yes"
job_crystal = AMSJob(settings=s_crystal, molecule=naph_crystal, name="naphthalene_crystal")
job_crystal.run();
[11.02|11:35:16] JOB naphthalene_crystal STARTED
[11.02|11:35:16] JOB naphthalene_crystal RUNNING
[11.02|11:35:36] JOB naphthalene_crystal FINISHED
[11.02|11:35:36] JOB naphthalene_crystal SUCCESSFUL
5) Lattice optimization results¶
After running the crystal job, you can extract the optimized cell lengths and angles.
lengths = naph_crystal.lattice.get_lengths(unit="angstrom")
angles = naph_crystal.lattice.get_angles(unit="degree")
print("Initial lengths:", lengths)
print("Initial angles:", angles)
Initial lengths: [8.0348 5.8899 8.565 ]
Initial angles: [ 90. 123.58999997 90. ]
opt_naph_crystal = job_crystal.results.get_main_system()
opt_naph_crystal.symmetrize() # clean up some numerical noise
lengths = opt_naph_crystal.lattice.get_lengths(unit="angstrom")
angles = opt_naph_crystal.lattice.get_angles(unit="degree")
print("Optimized lengths:", lengths)
print("Optimized angles:", angles)
Optimized lengths: [7.98578655 6.02392682 8.59150217]
Optimized angles: [ 90. 122.86278172 90. ]
6) Cohesive energy calculation¶
Use the same formula as the GUI tutorial:
E_coh = (2 * E_gas - E_crystal) / 2
E_gas = job_gas.results.get_energy(unit="kJ/mol")
E_crystal = job_crystal.results.get_energy(unit="kJ/mol")
E_coh = (2 * E_gas - E_crystal) / 2
print(f"E_gas = {E_gas:.2f} kJ/mol")
print(f"E_crystal = {E_crystal:.2f} kJ/mol")
print(f"E_coh = {E_coh:.2f} kJ/mol")
E_gas = -11584.06 kJ/mol
E_crystal = -23346.80 kJ/mol
E_coh = 89.34 kJ/mol
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## M3GNet-UP-2022 cohesive energy (naphthalene)
#
# This PLAMS workflow mirrors the GUI tutorial and uses the same structures, settings, and steps:
#
# - Gas-phase naphthalene geometry optimization
# - Crystal naphthalene lattice + geometry optimization
# - D3(BJ) dispersion add-on
# - Cohesive energy calculation from the two energies
# ### 1) Gas-phase naphthalene structure
from scm.base import ChemicalSystem
from scm.plams import view
naph_gas = ChemicalSystem(
"""
System
Atoms
C -2.43705000 0.70853000 0.00000000
C -1.24580000 1.40435000 0.00000000
C -1.24580000 -1.40435000 0.00000000
C 0.00000000 0.71830000 0.00000000
C -2.43705000 -0.70853000 0.00000000
C 0.00000000 -0.71830000 0.00000000
C 1.24580000 1.40435000 0.00000000
C 2.43705000 0.70853000 0.00000000
C 2.43705000 -0.70853000 0.00000000
C 1.24580000 -1.40435000 0.00000000
H -1.24407000 2.49615000 0.00000000
H -3.38504000 1.24787000 0.00000000
H -3.38504000 -1.24787000 0.00000000
H -1.24407000 -2.49615000 0.00000000
H 1.24407000 -2.49615000 0.00000000
H 3.38504000 -1.24787000 0.00000000
H 3.38504000 1.24787000 0.00000000
H 1.24407000 2.49615000 0.00000000
End
End
"""
)
view(naph_gas, direction="along_pca3", guess_bonds=True, width=200, height=200, picture_path="picture1.png")
# ### 2) Shared M3GNet settings (maps to GUI panels)
#
# These settings correspond to:
#
# - **Task → Geometry Optimization**
# - **Model → M3GNet-UP-2022**
# - **Details → Engine Add-ons → D3 Dispersion → Yes**
# - Optional: **Details → Technical → Device → cuda:0**
from scm.plams import Settings
m3gnet_settings = Settings()
m3gnet_settings.runscript.nproc = 1
m3gnet_settings.input.MLPotential.Model = "M3GNet-UP-2022"
# D3(BJ) dispersion engine add-on
m3gnet_settings.input.ams.EngineAddons.D3Dispersion.Enabled = "Yes"
# Optional device setting for GPU or CPU
# m3gnet_settings.input.MLPotential.Device = "cuda:0" # is determined automatically by default
# ### 3) Gas-phase naphthalene (geometry optimization)
#
# Matches the GUI steps for the gas-phase molecule.
from scm.plams import AMSJob
s_gas = m3gnet_settings.copy()
s_gas.input.ams.Task = "GeometryOptimization"
job_gas = AMSJob(settings=s_gas, molecule=naph_gas, name="naphthalene_gas")
job_gas.run()
# ### 4) Crystalline naphthalene (lattice + geometry optimization)
#
# Matches the GUI steps for the crystal, including **Optimize lattice → Yes**.
naph_crystal = ChemicalSystem(
"""
System
Atoms
C -0.87388368 0.11073012 2.36589887
C 0.15273024 3.05568012 4.76889870
C 4.17013024 5.77916988 4.76889870
C 3.14351632 2.83421988 2.36589887
H -1.09410897 0.35103804 3.23705766
H 0.37295554 3.29598804 3.89773991
H 4.39035554 5.53886196 3.89773991
H 2.92329103 2.59391196 3.23705766
C -0.12579372 0.96653259 1.59034638
C -0.59535971 3.91148259 5.54445119
C 3.42204029 4.92336741 5.54445119
C 3.89160628 1.97841741 1.59034638
H 0.15997330 1.77874980 1.94280538
H -0.88112673 4.72369980 5.19199219
H 3.13627327 4.11115020 5.19199219
H 4.17737330 1.16620020 1.94280538
C 0.21357858 0.61726152 0.25185835
C -0.93473202 3.56221152 6.88293921
C 3.08266798 5.27263848 6.88293921
C 4.23097858 2.32768848 0.25185835
C -3.75675609 1.48307682 6.55901941
C 3.03560266 4.42802682 0.57577816
C 7.05300266 4.40682318 0.57577816
C 0.26064391 1.46187318 6.55901941
H -3.47488266 2.30353989 6.89435489
H 2.75372923 5.24848989 0.24044268
H 6.77112923 3.58636011 0.24044268
H 0.54251734 0.64141011 6.89435489
C 6.72508585 4.75845021 1.84862605
C 0.58856072 1.81350021 5.28617152
C -3.42883928 1.13144979 5.28617152
C 2.70768585 4.07639979 1.84862605
H 6.21662119 4.18477395 2.37588759
H 1.09702538 1.23982395 4.75890998
H -2.92037462 1.70512605 4.75890998
H 2.19922119 4.65007605 2.37588759
End
Lattice
8.03480000 0.00000000 0.00000000
0.00000000 5.88990000 0.00000000
-4.73855343 0.00000000 7.13479757
End
End
"""
)
naph_crystal_supercell = naph_crystal.make_supercell((2, 2, 2)) # for visualization
view(naph_crystal_supercell, direction="tilt_y", guess_bonds=True, width=200, height=200, picture_path="picture2.png")
s_crystal = m3gnet_settings.copy()
# Geometry optimization with lattice optimization enabled
s_crystal.input.ams.Task = "GeometryOptimization"
s_crystal.input.ams.GeometryOptimization.OptimizeLattice = "Yes"
job_crystal = AMSJob(settings=s_crystal, molecule=naph_crystal, name="naphthalene_crystal")
job_crystal.run()
# ### 5) Lattice optimization results
#
# After running the crystal job, you can extract the optimized cell lengths and angles.
lengths = naph_crystal.lattice.get_lengths(unit="angstrom")
angles = naph_crystal.lattice.get_angles(unit="degree")
print("Initial lengths:", lengths)
print("Initial angles:", angles)
opt_naph_crystal = job_crystal.results.get_main_system()
opt_naph_crystal.symmetrize() # clean up some numerical noise
lengths = opt_naph_crystal.lattice.get_lengths(unit="angstrom")
angles = opt_naph_crystal.lattice.get_angles(unit="degree")
print("Optimized lengths:", lengths)
print("Optimized angles:", angles)
# ### 6) Cohesive energy calculation
#
# Use the same formula as the GUI tutorial:
#
# E_coh = (2 * E_gas - E_crystal) / 2
E_gas = job_gas.results.get_energy(unit="kJ/mol")
E_crystal = job_crystal.results.get_energy(unit="kJ/mol")
E_coh = (2 * E_gas - E_crystal) / 2
print(f"E_gas = {E_gas:.2f} kJ/mol")
print(f"E_crystal = {E_crystal:.2f} kJ/mol")
print(f"E_coh = {E_coh:.2f} kJ/mol")