Multiple molecules multiple methods¶
The AMS-GUI can handle multiple molecules, or multiple methods, or both, with one relatively simple setup.
Multiple molecules¶
With AMSinput you can set up a calculation, and then run your set up for multiple molecules automatically. At run time the molecules are then taken from an .sdf file that has the same name as your .ams file (except for the extension obviously). This SDF file may come from somewhere else, or you can actually create and edit multiple molecules inside AMSinput.
After the calculation the result is a new .sdf file. That .sdf file is a standard SDF file, with the calculated energy in the title for each molecule. It also contains references to the calculation results for the individual molecules. And it may contain some extra information per molecule (depending on the calculation).
As an example, let’s calculate the H-NMR spectra for methane and ethane with one single set-up.
Set up methane and ethane in AMSinput¶
C 0.00000000 0.00000000 0.00000000
H 0.63290000 -0.63290000 -0.63290000
H -0.63290000 0.63290000 -0.63290000
H 0.63290000 0.63290000 0.63290000
H -0.63290000 -0.63290000 0.63290000
Next we will add an extra molecule (ethane) to AMSinput:
C 0.00000000 0.00000000 0.76580000
C 0.00000000 0.00000000 -0.76580000
H -0.88670000 0.51190000 1.16680000
H 0.88670000 0.51190000 1.16680000
H 0.00000000 -1.02390000 1.16680000
H 0.00000000 1.02390000 -1.16680000
H -0.88670000 -0.51190000 -1.16680000
H 0.88670000 -0.51190000 -1.16680000
H-NMR calculation¶
Next we will set up the calculation of an H-NMR:
In the AMSjobs window you should see the job running. Also note that it gives you the information that the job uses the nmr.sdf file. When you open the job you will see the results for two different molecules in the .results folder:
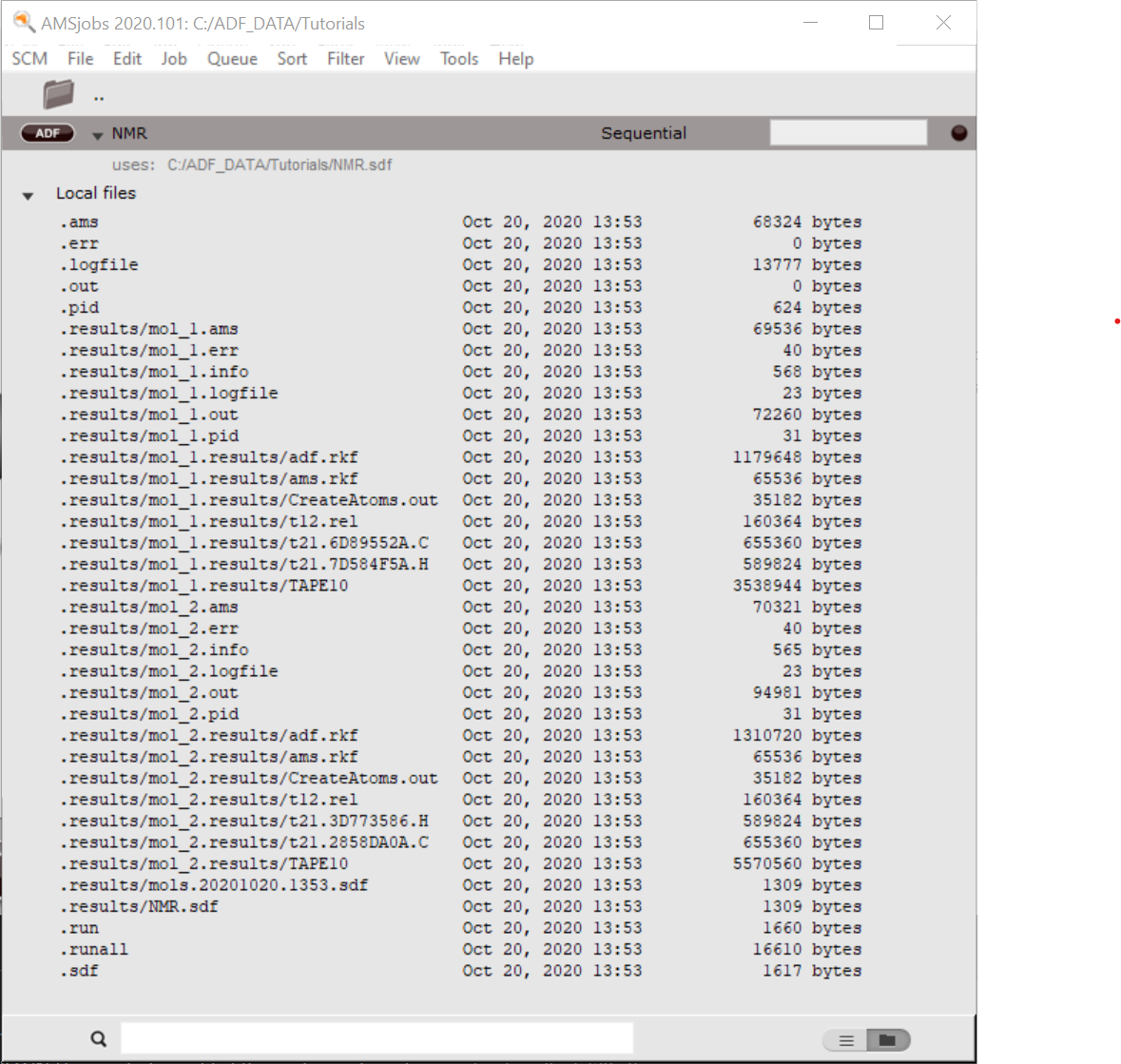
Note that the results of the calculated NMR spectra will depend on the geometries for methane and ethane, on the used basis set, XC functional, etcetera. See also ADF FAQ: What settings are recommended for NMR calculations?
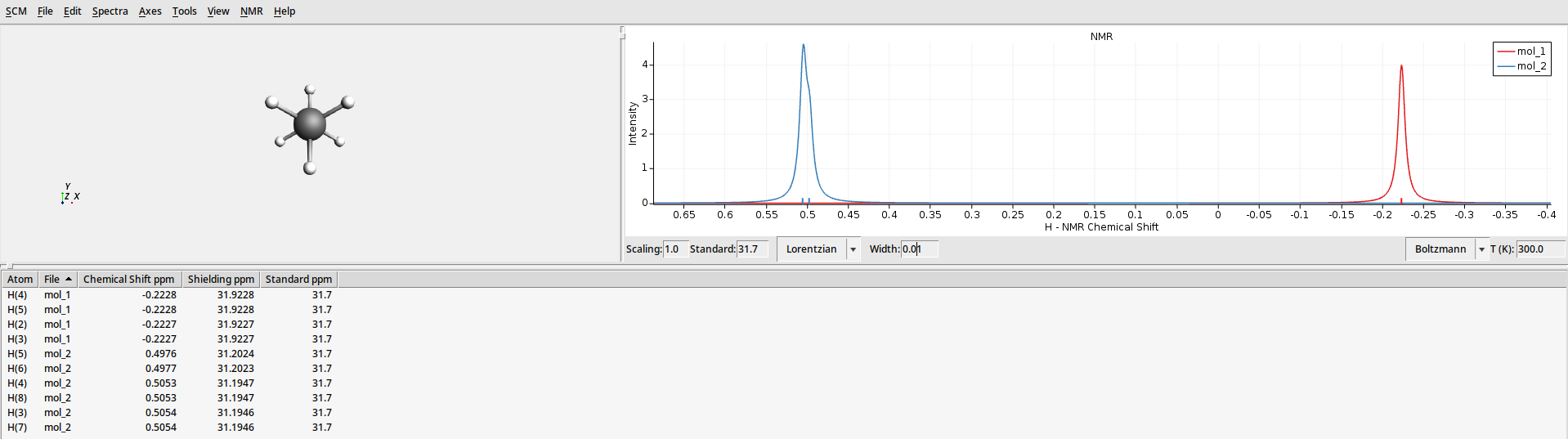
Use AMSspectra to see the calculated NMR spectra. When opening the job, it will show an averaged spectrum. Use the View menu to adjust what curves to see:
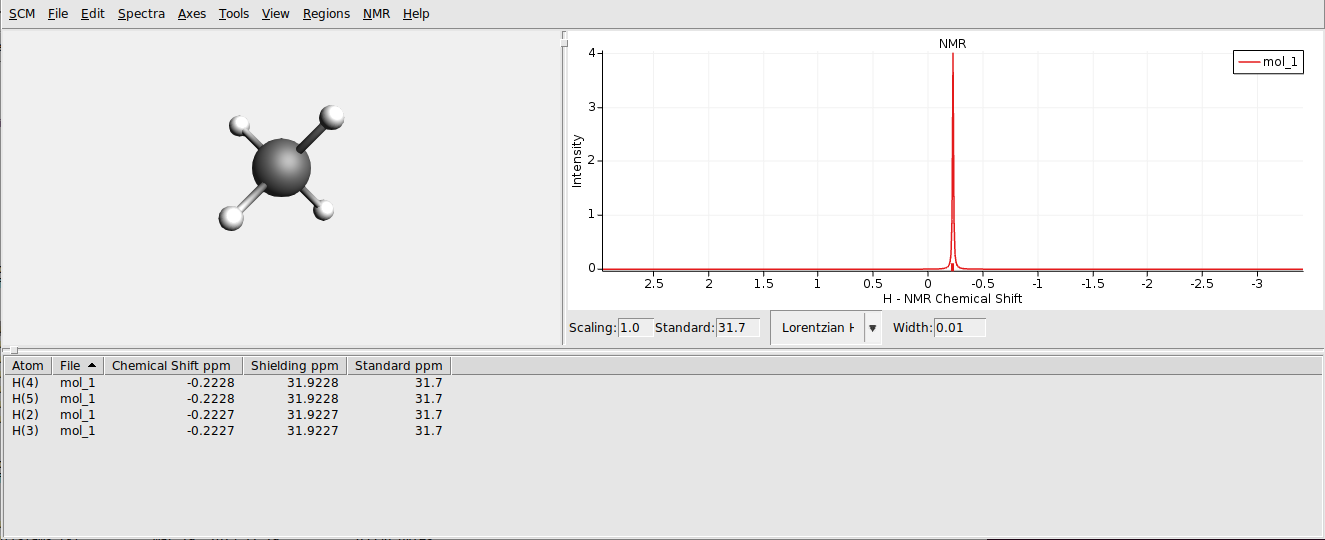
You can also open the result files for the individual calculations:
Multiple Methods¶
You can also set up a series of calculations with different methods, and run them automatically one after each other. The geometries will automatically be passed from one job to the next.
As examples we first show how to set up a series like: DFTB optimization, ADF optimization and ADF UV/Vis spectrum for the optimized structure. We will then run this set up twice: once for the specified molecule, and once for a set of molecules.
Set up a series of calculations¶
The first step in the series is a DFTB calculation on a molecule, we will use Acetone as an example:
The second step in the series is an ADF geometry optimization of the optimized structure from the first step:
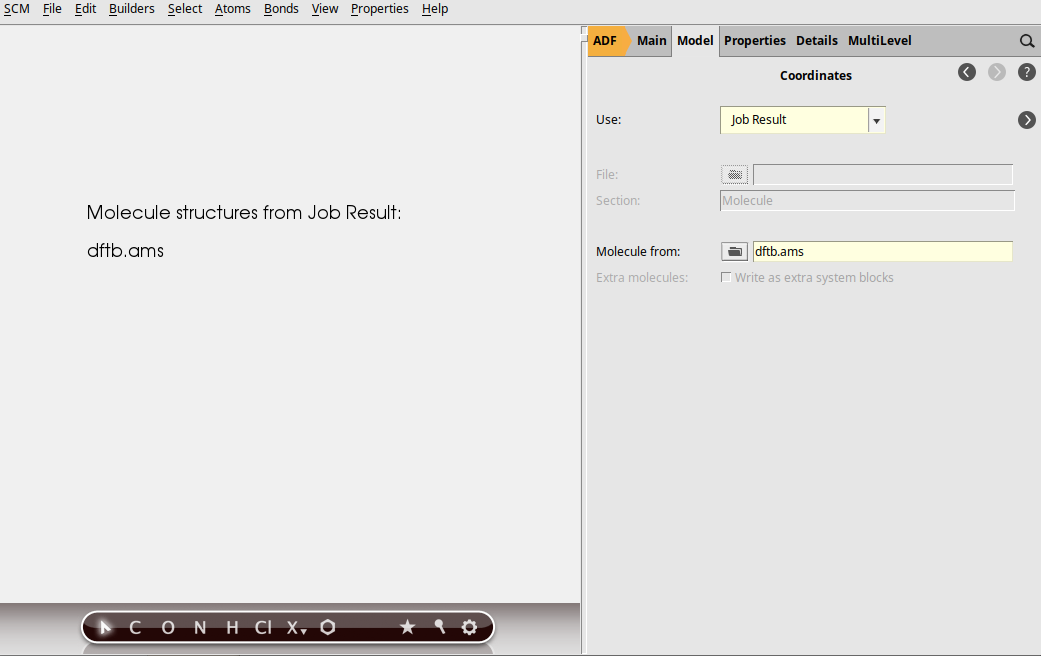
We now use the Job Result option in the Coordinates panel: this will make sure that the selected job is run first, and that the resulting geometry of that job will be used as input geometry for the current calculation.
The third step in the series is an ADF UV/Vis calculation for the ADF-optimized structures:
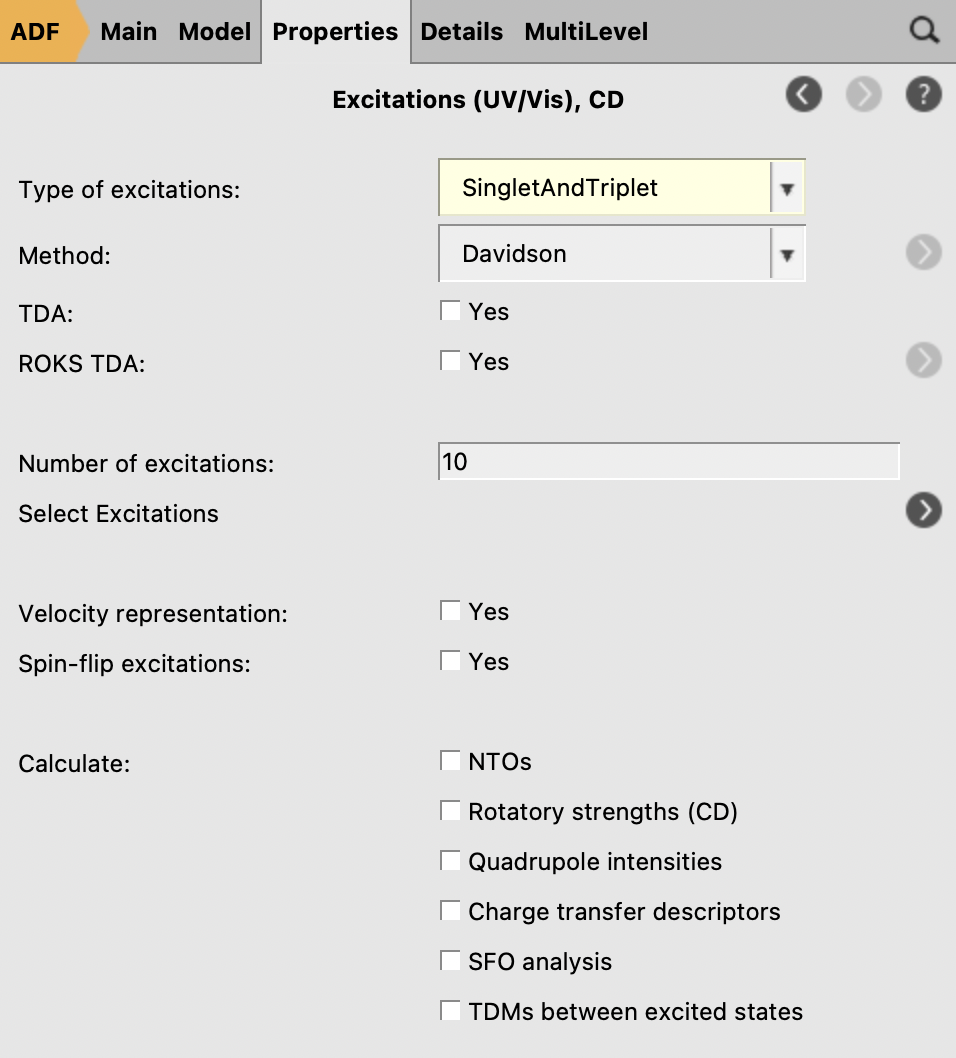
Run a series of calculations for a single molecule¶
To run the just created series of calculation all we need to do is to start the final job (adf-uvvis). AMSjobs will detect that it uses results of other jobs, and start those jobs automatically:
In the AMSjobs window you can see on which jobs the selected job depends:
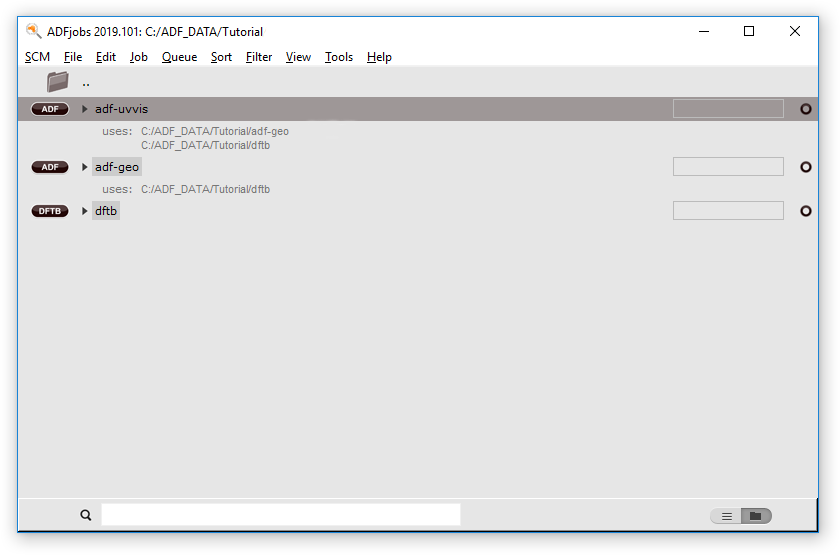
In AMSjobs you should now see all the jobs in the job series running, one by one, in the proper order. If you wish you can examine the results of each job step as you would for normal independent jobs.
Lets now just check the final UV/Vis spectrum:
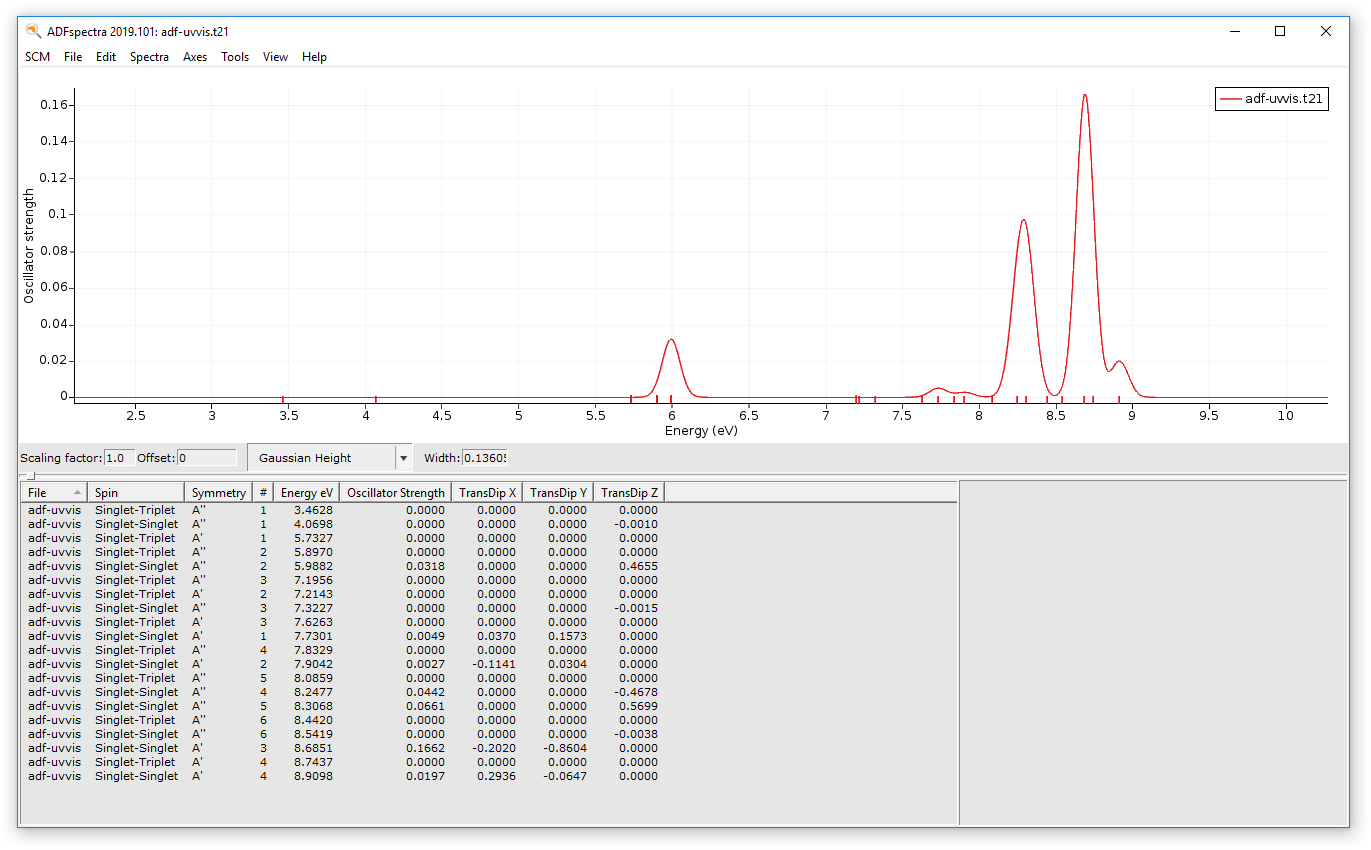
Remember that in the adf-uvvis job no molecule was specified, the geometry has been imported from the result of the adf-geo job. And that job in turn got the geometry from the dftb job.
Create an SDF file, and Run a series of calculations for a set of molecules¶
The series of calculations in the previous steps can also be run with molecules from a .sdf file as input. Any SDF file can be used, for example the earlier sdf files from this tutorial.
In this case we will make a new SDF file just to show how to do that as well. First we make two .ams files, one for benzene and one for water:
Next we make an SDF file containing the two molecules we just made:
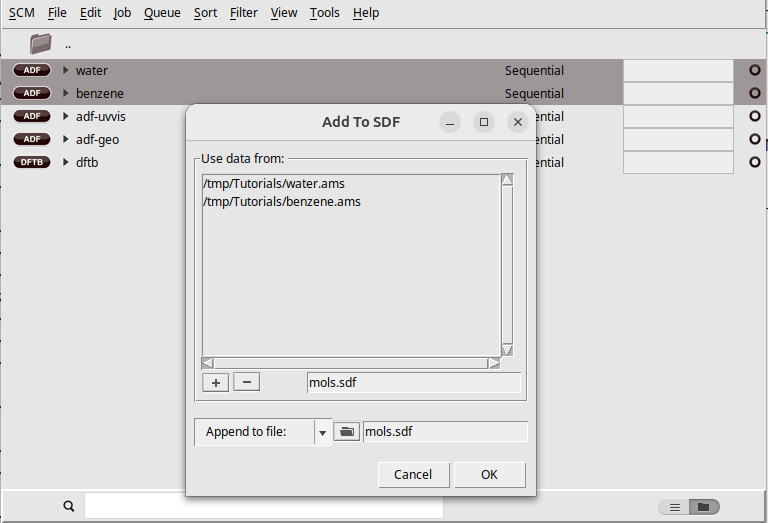
A new SDF file with the specified name will be created, containing the structures you selected. You can also add extra molecules to the .sdf file using the same method (just select an existing .sdf file instead of a new one). You can also include structures resulting from calculation results you have (.t21, .rkf files etc), or even from simple .xyz or .mol files and so on.
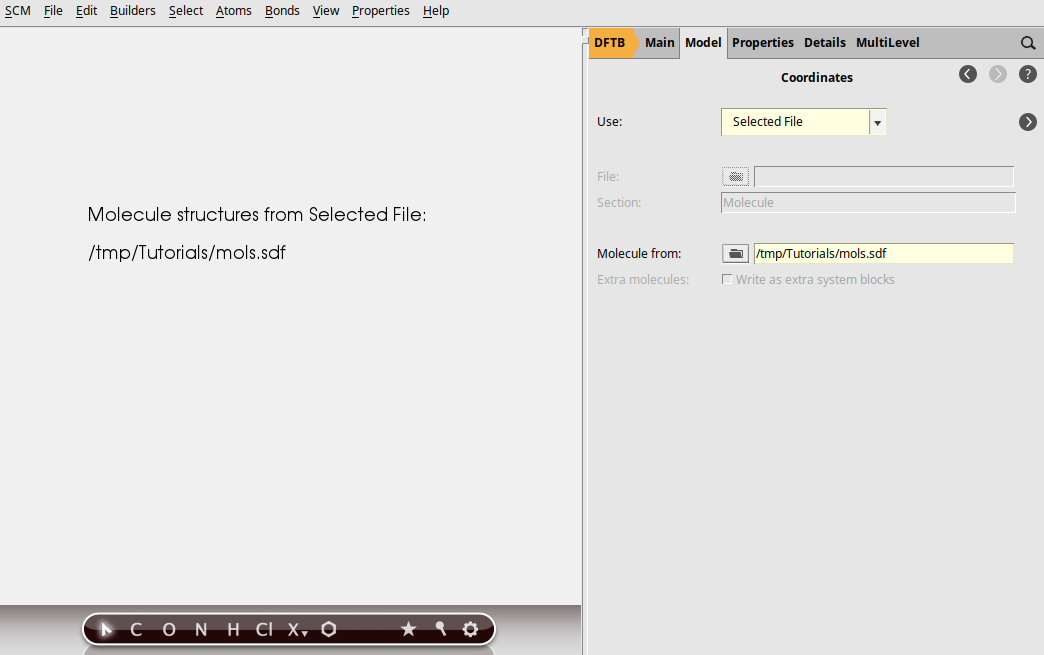
Next we want to run the series of jobs (DFTB - ADF geometry optimization - ADF UV/Vis) for all molecules in the mols.sdf file. In the first job of the series (DFTB) specify to use molecules from the .sdf file:
You should see all jobs running again in AMSjobs, but now multiple times. In the .results directories you will find the results for the individual jobs, and the final geometries are always collected in new .sdf files.
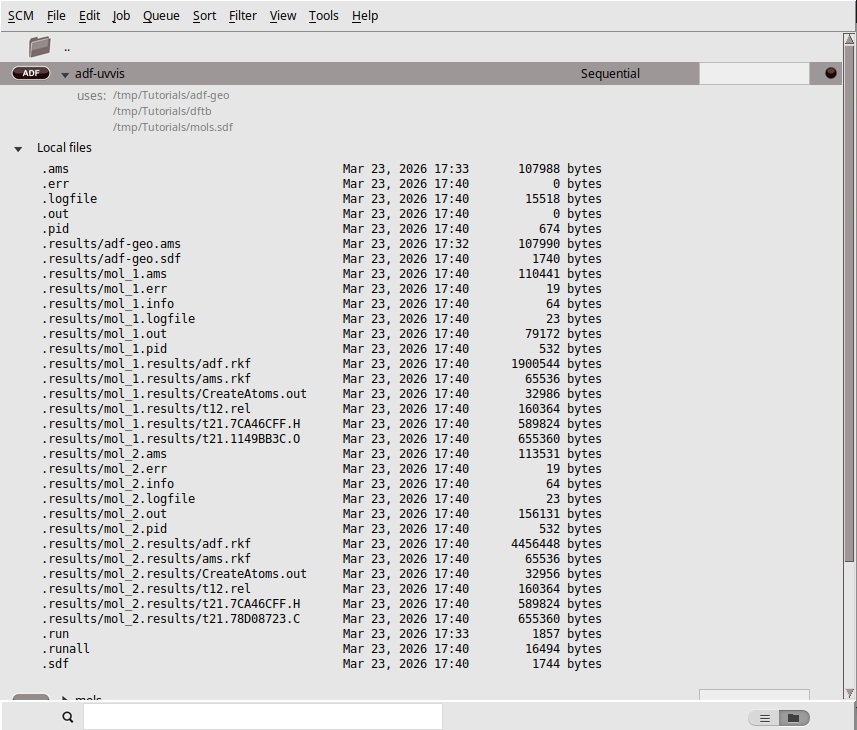
You can see the average or individual spectra via AMSspectra, or the geometries using AMSmovie:
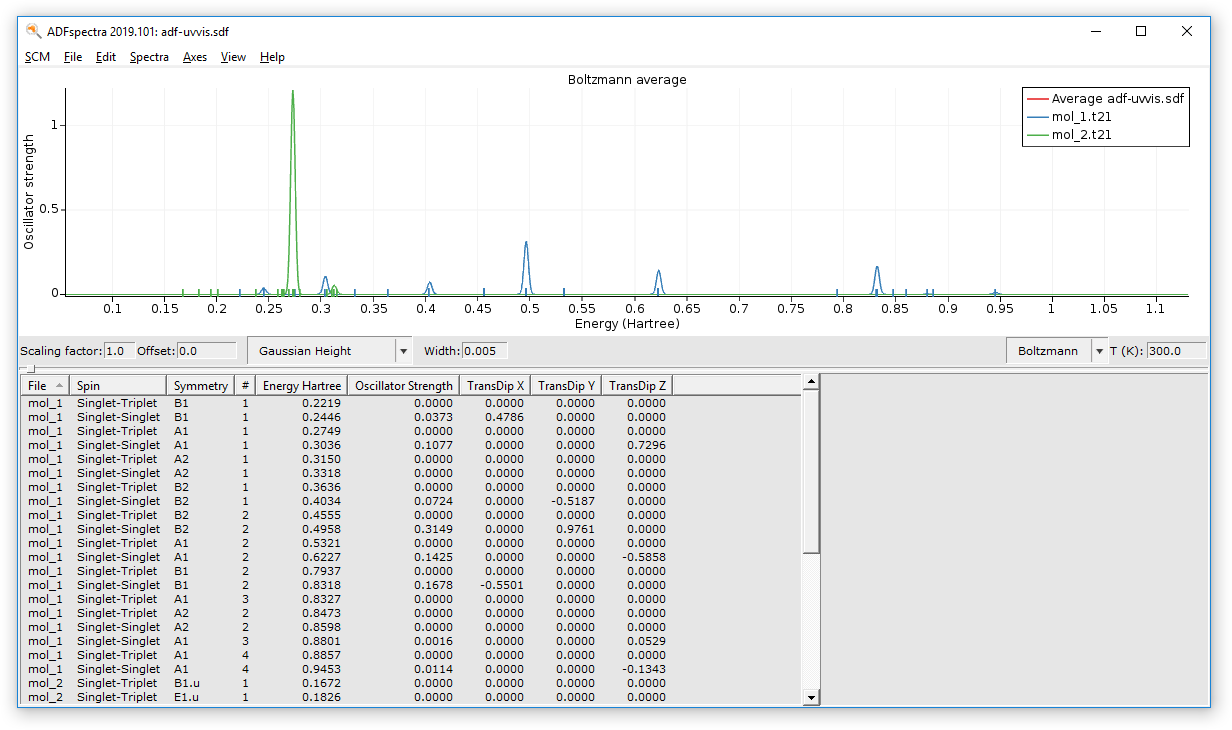
Such a multi molecule job can also be inspected with amsmovie
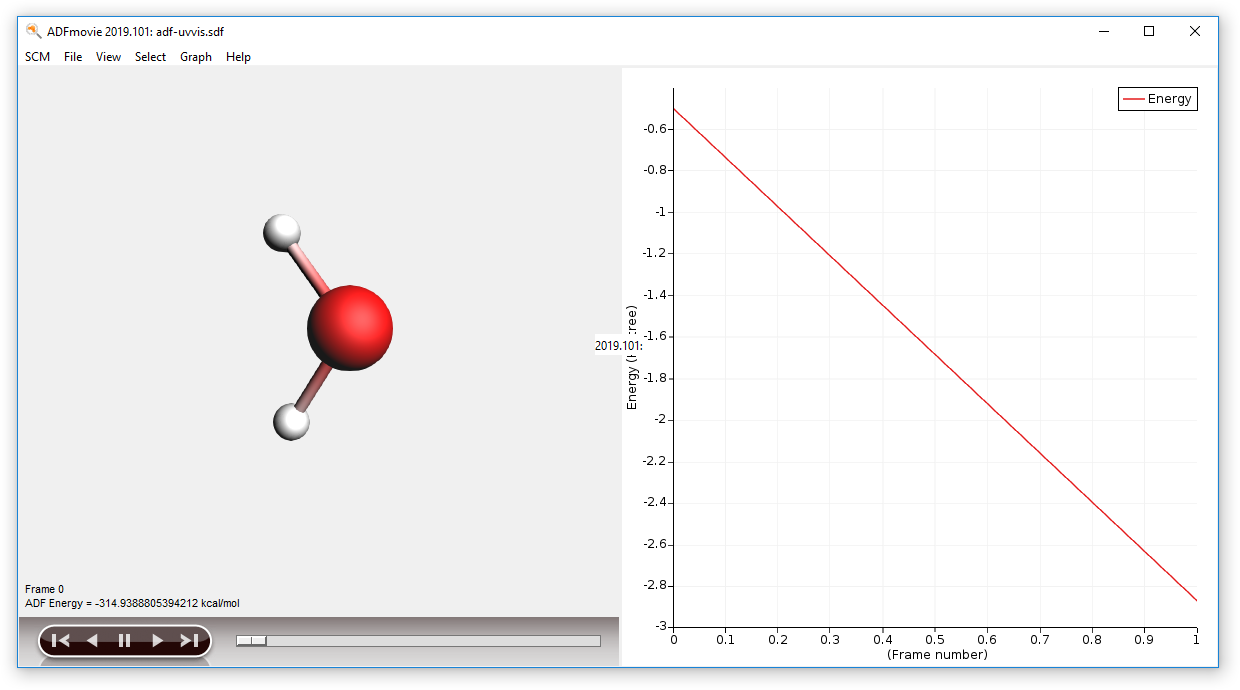
Here each frame corresponds to the final energy of the optimized molecules (two in this example). Comparing energies of different molecules is not very useful, but this feature makes sense when considering conformers.