Powerful molecular DFT to understand chemistry
Learn more
Experience what the Amsterdam Modeling Suite can do for you!
Advance your research in Chemistry, Materials or Engineering.

“What I really like about the Amsterdam Modeling Suite is that the programs were clearly written by chemists for dealing with real chemical problems. A great suite of programs!”
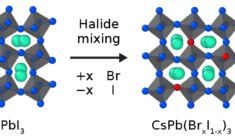
Key concepts: Parametrization, perovskite, ReaxFF
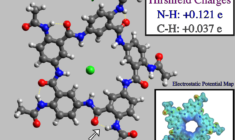
Key concepts: ADF, bonding analysis
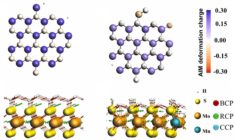
Key concepts: BAND, bonding analysis, catalysis, materials science, nanoscience, periodic DFT, Relativistic DFT