3.4. ReaxFF: Training set for cobalt¶
The cobalt training set documented here was used in the parametrization of the Cobalt force field, Co.ff, published in “Development of a Transferable Reactive Force Field for Cobalt” by M. R. LaBrosse et al. Within the publication the choice of training set entries is discussed from a scientific standpoint.
This page only discusses the training set, not the parametrization.
Contents:
The job_collection.yaml and training_set.yaml files can be found in the
directory $AMSHOME/scripting/scm/params/examples/import_old_ReaxFF/Co/converted_to_params,
where $AMSHOME is the AMS installation directory.
In the ParAMS GUI, select File → Open and browse to the job_collection.yaml file.
3.4.1. Weighting of individual entries¶
To estimate the quality of a given set of ReaxFF parameters with respect to the training data a sum of squared errors loss function (or objective function, or error function) is defined:
where the sum runs over all training set entries. Each difference between the reference property (xi,ref) and the ReaxFF value (xi,ReaxFF) is weighted individually via the weight wi and σi.
For ReaxFF parametrization up to AMS2021, only the σ value was used. In ParAMS, both w and σ are used. You can modify one or both of them, depending on your preference. For more details, see Sigma vs. weight: What is the difference?.
Note
The value of this loss function is the quantity that is minimized by the optimization algorithms used for the force field fitting. It is therefore important to choose the weightings such that there is no unwanted bias towards one or the other entry.
In total the Co training set contains 144 entries all of which are energies. The training data is distributed as follows:
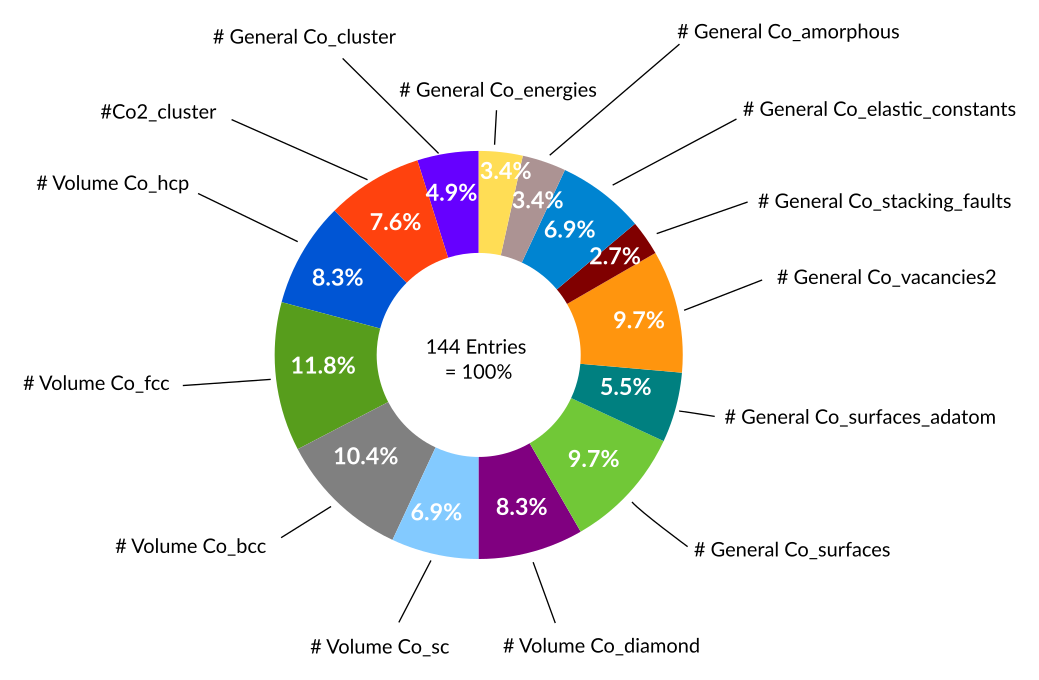
The contribution of each entry to the overall error function can to some extent be visualized using the weightings of the error function.
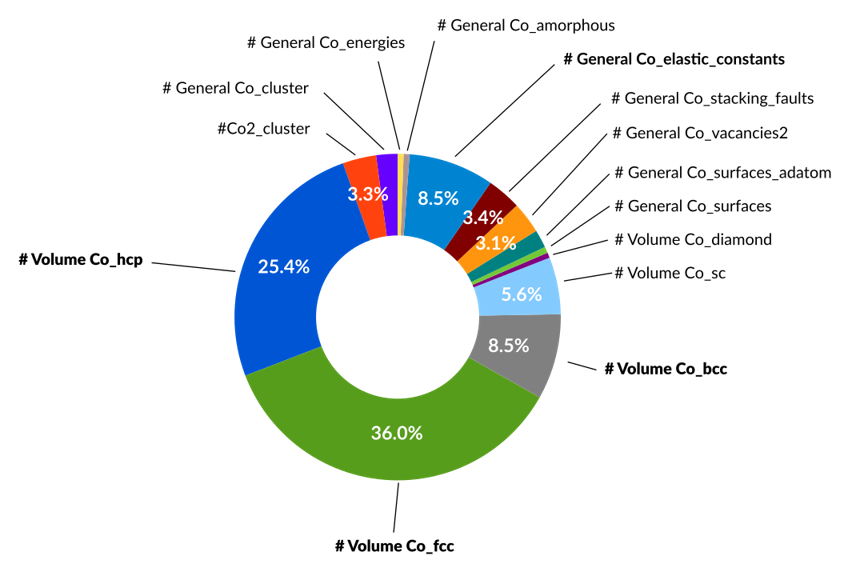
For example, the most stable phases hcp and fcc are given much higher weights during the optimization (25.4% and 36.0%) than the less favorable diamond phase (0.5%). In practice one would use the breakdown of the error function (Loss contribution) to finetune the weights of the loss function, which is probably how the above weightings were set too.
3.4.2. General energies, Cluster models and the Co₂ dimer¶
The energy differences between optimized cubic phases are included in the training data
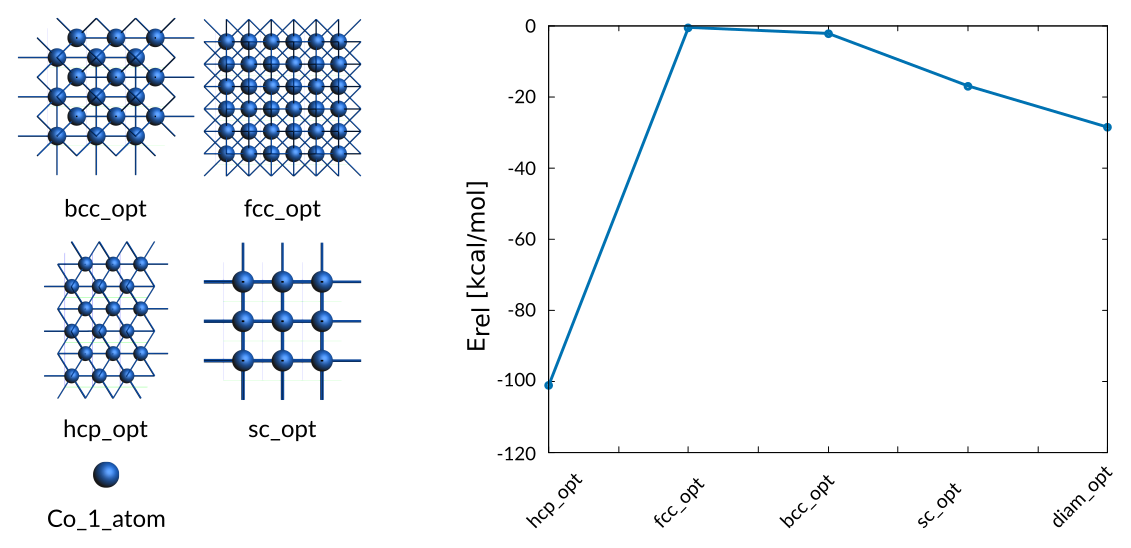
Weight Sigma Reference_value Expression
6.300 1.255 -101.300 kcal/mol +energy("hcp_opt")/2-energy("Co_1_atom")/1
3937.683 1.255 -0.520 kcal/mol +energy("hcp_opt")/2-energy("fcc_opt")/4
157.507 1.255 -2.210 kcal/mol +energy("hcp_opt")/2-energy("bcc_opt")/2
1.575 1.255 -17.000 kcal/mol +energy("hcp_opt")/2-energy("sc_opt")/1
0.394 1.255 -28.600 kcal/mol +energy("hcp_opt")/2-energy("diam_opt")/8
In addition cohesive energies for a set of small clusters of sizes 2, 3, 4, 5, 6, 8, and 13 atoms are included
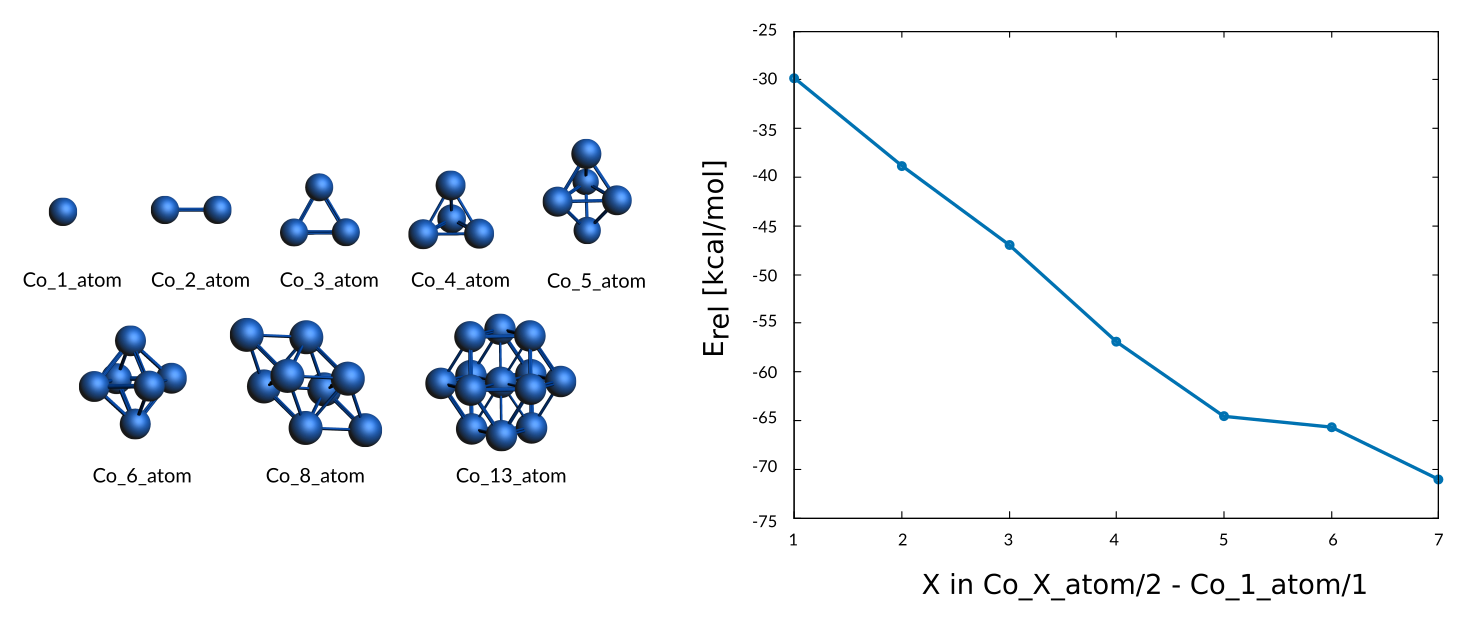
Weight Sigma Reference_value Expression
0.394 1.255 -29.790 kcal/mol +energy("Co_2_atom")/2-energy("Co_1_atom")/1
0.394 1.255 -38.800 kcal/mol +energy("Co_3_atom")/3-energy("Co_1_atom")/1
0.394 1.255 -46.940 kcal/mol +energy("Co_4_atom")/4-energy("Co_1_atom")/1
0.394 1.255 -56.910 kcal/mol +energy("Co_5_atom")/5-energy("Co_1_atom")/1
0.394 1.255 -64.580 kcal/mol +energy("Co_6_atom")/6-energy("Co_1_atom")/1
0.394 1.255 -65.680 kcal/mol +energy("Co_8_atom")/8-energy("Co_1_atom")/1
0.394 1.255 -71.060 kcal/mol +energy("Co_13_atom")/13-energy("Co_1_atom")/1
A scan of the bond stretch for the Co₂ dimer is included as well
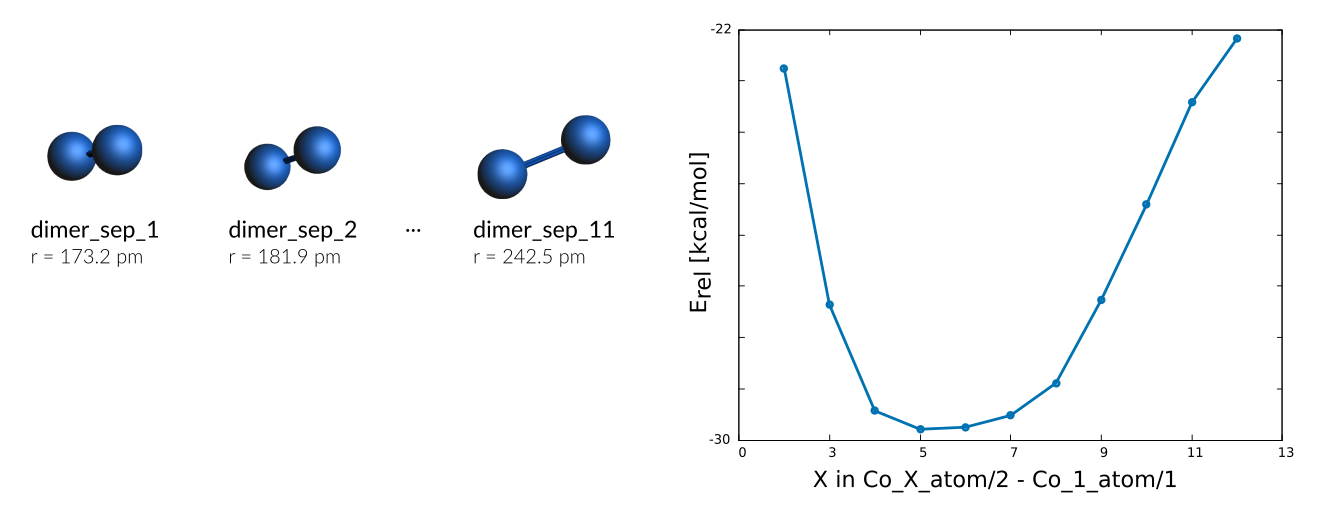
Weight Sigma Reference_value Expression
0.063 1.255 -22.750 kcal/mol +energy("dimer_sep_1")/2-energy("Co_1_atom")/1
0.063 1.255 -27.360 kcal/mol +energy("dimer_sep_2")/2-energy("Co_1_atom")/1
0.063 1.255 -29.430 kcal/mol +energy("dimer_sep_3")/2-energy("Co_1_atom")/1
0.063 1.255 -29.790 kcal/mol +energy("dimer_sep_4")/2-energy("Co_1_atom")/1
0.063 1.255 -29.750 kcal/mol +energy("dimer_sep_5")/2-energy("Co_1_atom")/1
0.063 1.255 -29.520 kcal/mol +energy("dimer_sep_6")/2-energy("Co_1_atom")/1
0.063 1.255 -28.890 kcal/mol +energy("dimer_sep_7")/2-energy("Co_1_atom")/1
0.063 1.255 -27.270 kcal/mol +energy("dimer_sep_8")/2-energy("Co_1_atom")/1
0.063 1.255 -25.410 kcal/mol +energy("dimer_sep_9")/2-energy("Co_1_atom")/1
0.063 1.255 -23.420 kcal/mol +energy("dimer_sep_10")/2-energy("Co_1_atom")/1
0.063 1.255 -22.170 kcal/mol +energy("dimer_sep_11")/2-energy("Co_1_atom")/1
Not surprisingly the dimer entries of the bond stretch are single point calculations. See how to import your own bond scans into ParAMS.
3.4.3. Description of crystalline phases¶
The training set includes equation of state (EOS) curves (energy-volume curves) for the following crystalline phases
hcp
fcc
bcc
sc
diamond
The curves were generated by performing a complete relaxation with a fixed cell volume. The according energies are defined per-atom and set relative to the structure with the lowest energy. For example, the EOS for the hcp and diamond phases are defined as follows:
hcp phase
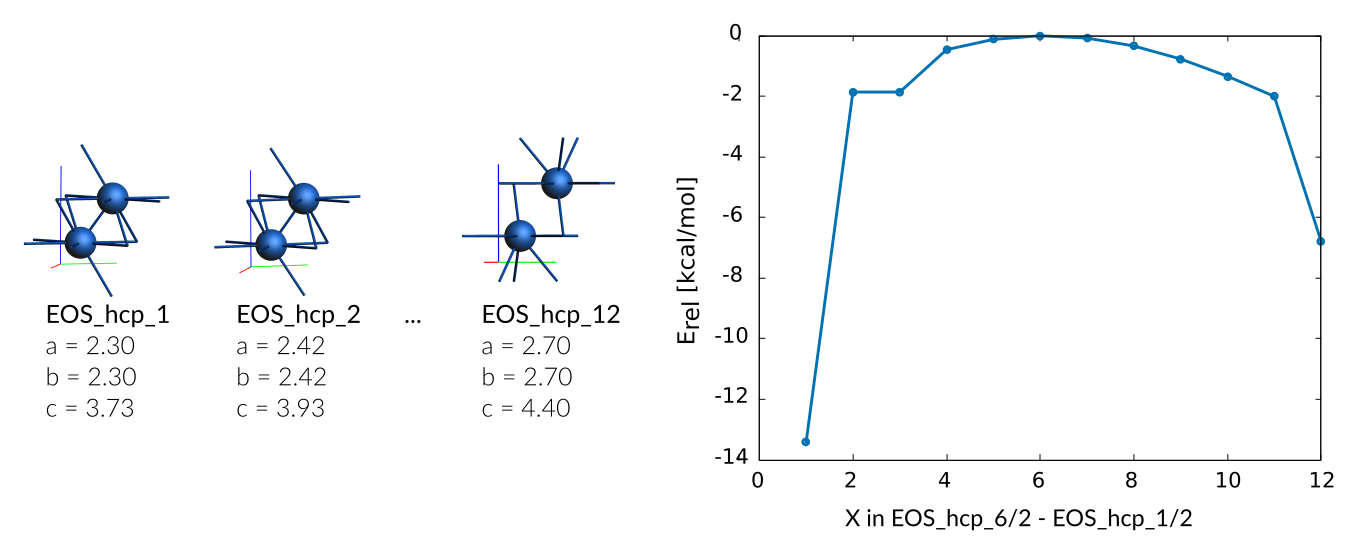
Weight Sigma Reference_value Expression
9.844 1.255 -13.420 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_1")/2
39.377 1.255 -1.860 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_2")/2
157.507 1.255 -1.040 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_3")/2
157.507 1.255 -0.460 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_4")/2
157.507 1.255 -0.120 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_5")/2
157.507 1.255 -0.010 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_6")/2
157.507 1.255 -0.080 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_7")/2
157.507 1.255 -0.340 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_8")/2
157.507 1.255 -0.770 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_9")/2
157.507 1.255 -1.340 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_10")/2
39.377 1.255 -2.000 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_11")/2
9.844 1.255 -6.790 kcal/mol +energy("EOS_hcp_6")/2-energy("EOS_hcp_12")/2
diamond phase
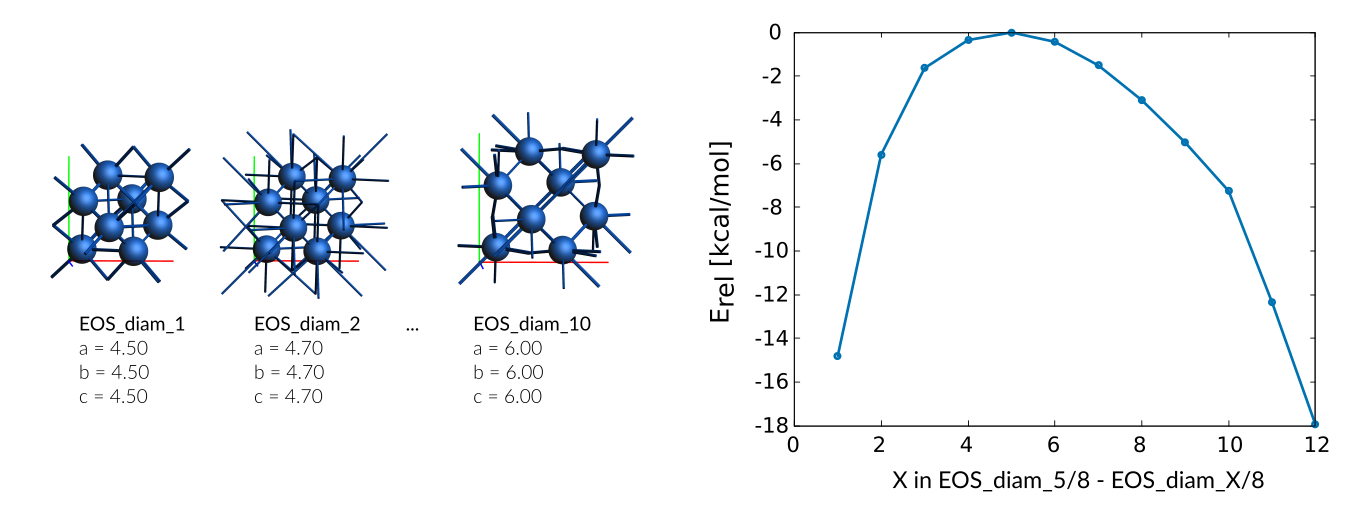
Weight Sigma Reference_value Expression
0.025 1.255 -14.800 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_1")/8
0.025 1.255 -5.590 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_2")/8
0.025 1.255 -1.630 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_3")/8
0.025 1.255 -0.350 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_4")/8
0.025 1.255 -0.010 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_5")/8
0.025 1.255 -0.430 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_6")/8
0.025 1.255 -1.510 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_7")/8
0.025 1.255 -3.100 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_8")/8
0.025 1.255 -5.040 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_9")/8
0.025 1.255 -7.260 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_10")/8
0.025 1.255 -12.350 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_11")/8
0.025 1.255 -17.950 kcal/mol +energy("EOS_diam_5")/8-energy("EOS_diam_12")/8
3.4.4. Description of Co-surfaces¶
The training set contains several surfaces for which the training values are modified surface formation energies defined as the per atom energy of the surface relative to the energy per atom of the bulk phase (optimized hcp).
For the cubic surfaces (fcc,bcc,sc) both low-Miller and high-Miller surfaces are included. The (0001) surface has been added for the hcp phase only.
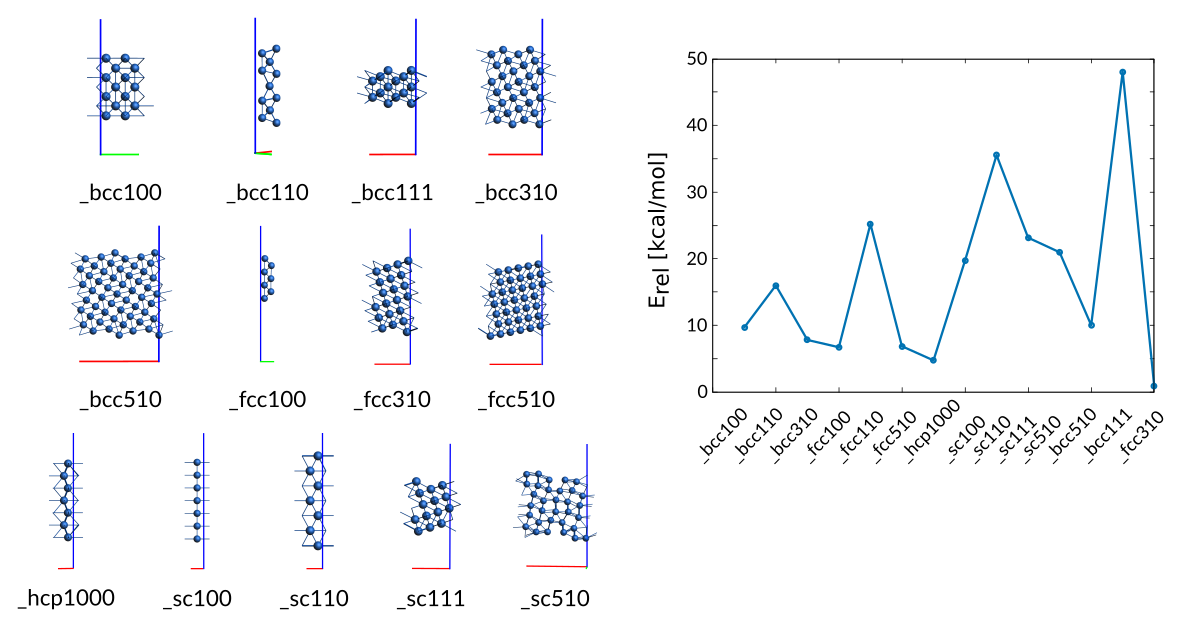
Weight Sigma Reference_value Expression
0.394 1.255 9.670 kcal/mol +energy("Surface_bcc100")/28-energy("hcp_opt")/2
0.394 1.255 15.970 kcal/mol +energy("Surface_bcc110")/11-energy("hcp_opt")/2
0.394 1.255 7.790 kcal/mol +energy("Surface_bcc310")/30-energy("hcp_opt")/2
157.507 1.255 6.690 kcal/mol +energy("Surface_fcc100")/14-energy("hcp_opt")/2
0.025 1.255 25.200 kcal/mol +energy("Surface_fcc110")/7-energy("hcp_opt")/2
0.394 1.255 6.840 kcal/mol +energy("Surface_fcc510")/37-energy("hcp_opt")/2
630.029 1.255 4.710 kcal/mol +energy("Surface_hcp1000")/7-energy("hcp_opt")/2
0.005 1.255 19.720 kcal/mol +energy("Surface_sc100")/7-energy("hcp_opt")/2
0.005 1.255 35.550 kcal/mol +energy("Surface_sc110")/7-energy("hcp_opt")/2
0.005 1.255 23.140 kcal/mol +energy("Surface_sc111")/14-energy("hcp_opt")/2
0.005 1.255 20.950 kcal/mol +energy("Surface_sc510")/32-energy("hcp_opt")/2
0.000 1.255 10.000 kcal/mol +energy("Surface_bcc510")/54-energy("hcp_opt")/2
0.003 1.255 48.060 kcal/mol +energy("Surface_bcc111")/14-energy("hcp_opt")/2
0.025 1.255 0.810 kcal/mol +energy("Surface_fcc310")/21-energy("hcp_opt")/2
Tip
Creating various surfaces and bulk materials is easy with the GUI.
3.4.5. Adatoms¶
Since the migration of Cobalt atoms on various surfaces is essential for the forming of energetically favorable surfaces, the training set contains a variety of adatom structures (top, bridge, hollow sites) on a variety of surfaces (fcc, bcc, sc). All DFT references were optimized, to ensure that the adatom is located in a local minimum.
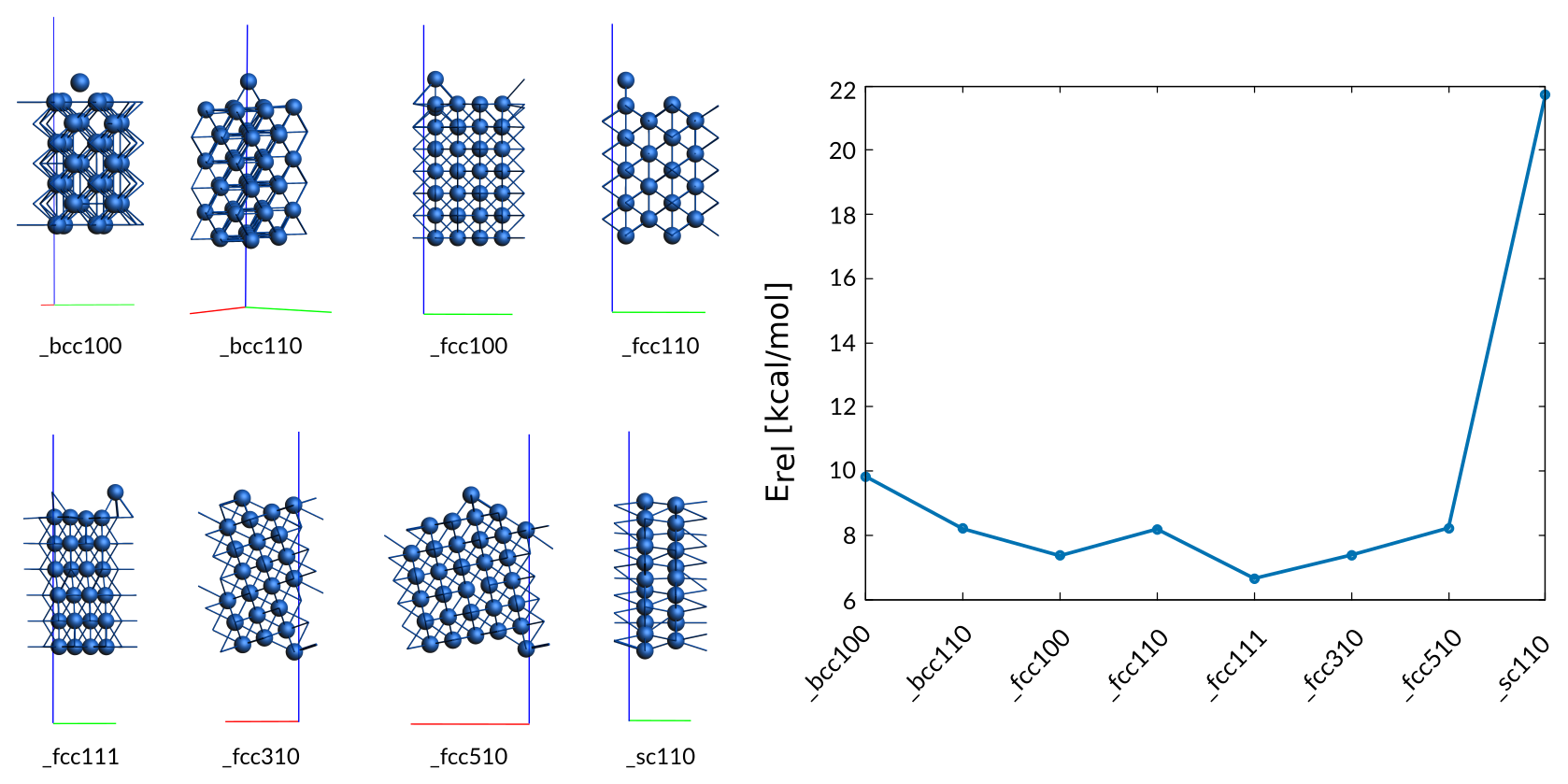
Weight Sigma Reference_value Expression
0.394 1.255 9.830 kcal/mol +energy("Surf_adatom_bcc100")/29-energy("hcp_opt")/2
0.394 1.255 8.200 kcal/mol +energy("Surf_adatom_bcc110")/49-energy("hcp_opt")/2
0.394 1.255 7.360 kcal/mol +energy("Surf_adatom_fcc100")/57-energy("hcp_opt")/2
0.394 1.255 8.190 kcal/mol +energy("Surf_adatom_fcc110")/37-energy("hcp_opt")/2
0.394 1.255 6.650 kcal/mol +energy("Surf_adatom_fcc111")/25-energy("hcp_opt")/2
0.394 1.255 7.380 kcal/mol +energy("Surf_adatom_fcc310")/43-energy("hcp_opt")/2
0.394 1.255 8.220 kcal/mol +energy("Surf_adatom_fcc510")/63-energy("hcp_opt")/2
0.063 1.255 21.740 kcal/mol +energy("Surf_adatom_sc110")/29-energy("hcp_opt")/2
3.4.6. Vacancies and defects¶
To consider vacancies and defects in bulk cobalt the training set contains
bulk fcc cobalt with missing Co atoms (vacancies)
amorphous bulk Co structures
stacking fault defects
The training set contains formation energies of 1−6 coalesced vacancies in fcc cobalt. As discussed in the paper, the training data shows that it is most energetically favorable to have two vacancies as nearest neighbors.
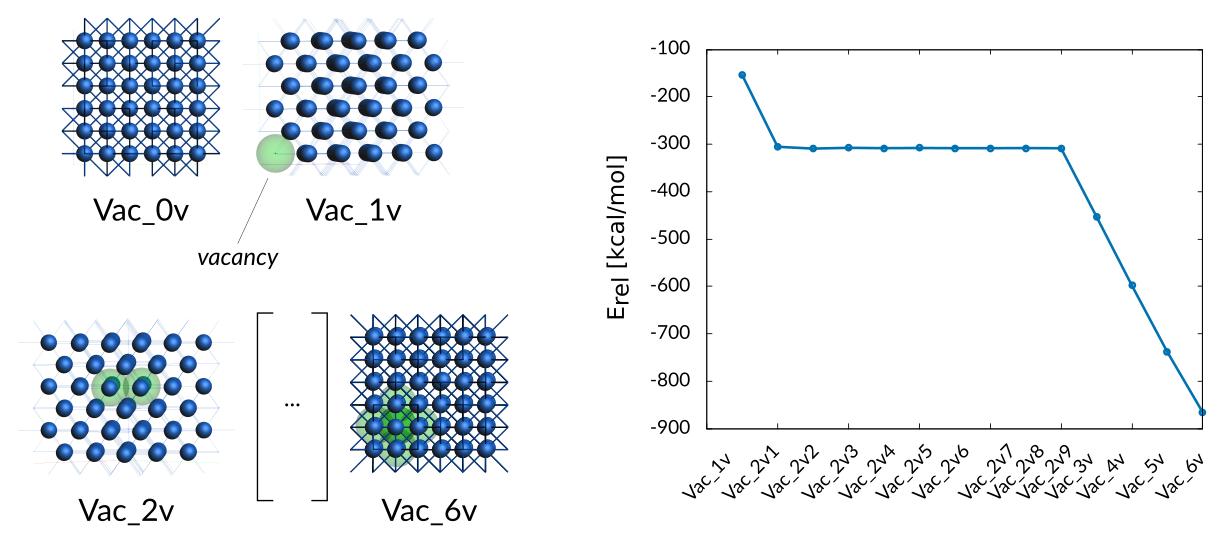
Weight Sigma Reference_value Expression
1.575 1.255 -154.090 kcal/mol +energy("Vac_0v")/1-energy("Vac_1v")/1-energy("Co_1_atom")/1
0.098 1.255 -305.260 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v1")/1-energy("Co_1_atom")/0.5
0.098 1.255 -309.220 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v2")/1-energy("Co_1_atom")/0.5
0.098 1.255 -307.460 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v3")/1-energy("Co_1_atom")/0.5
0.098 1.255 -308.340 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v4")/1-energy("Co_1_atom")/0.5
0.098 1.255 -307.880 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v5")/1-energy("Co_1_atom")/0.5
0.098 1.255 -308.270 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v6")/1-energy("Co_1_atom")/0.5
0.098 1.255 -308.330 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v7")/1-energy("Co_1_atom")/0.5
0.098 1.255 -308.000 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v8")/1-energy("Co_1_atom")/0.5
0.098 1.255 -308.370 kcal/mol +energy("Vac_0v")/1-energy("Vac_2v9")/1-energy("Co_1_atom")/0.5
0.098 1.255 -452.510 kcal/mol +energy("Vac_0v")/1-energy("Vac_3v")/1-energy("Co_1_atom")/0.33333333
0.098 1.255 -598.220 kcal/mol +energy("Vac_0v")/1-energy("Vac_4v")/1-energy("Co_1_atom")/0.25
0.098 1.255 -738.330 kcal/mol +energy("Vac_0v")/1-energy("Vac_5v")/1-energy("Co_1_atom")/0.2
0.098 1.255 -865.980 kcal/mol +energy("Vac_0v")/1-energy("Vac_6v")/1-energy("Co_1_atom")/0.16666667
Amorphous bulk Co was generated by running ab-initio NVT MD at 2500K of 108 atom fcc lattice. The snapshots from the trajectory are included as single point calculations in the training data.

Weight Sigma Reference_value Expression
0.025 1.255 14.130 kcal/mol +energy("Amorphous_1")/108-energy("hcp_opt")/2
0.025 1.255 10.350 kcal/mol +energy("Amorphous_2")/108-energy("hcp_opt")/2
0.025 1.255 8.480 kcal/mol +energy("Amorphous_3")/108-energy("hcp_opt")/2
0.025 1.255 7.270 kcal/mol +energy("Amorphous_4")/108-energy("hcp_opt")/2
0.098 1.255 8.500 kcal/mol +energy("Amorphous_5")/108-energy("hcp_opt")/2
Stacking fault energies for the cubic phases were created via a half-lattice offset in [100] direction. The stacking fault energy for the hcp phase is described via a layering transition from hcp(0001) to fcc(111).
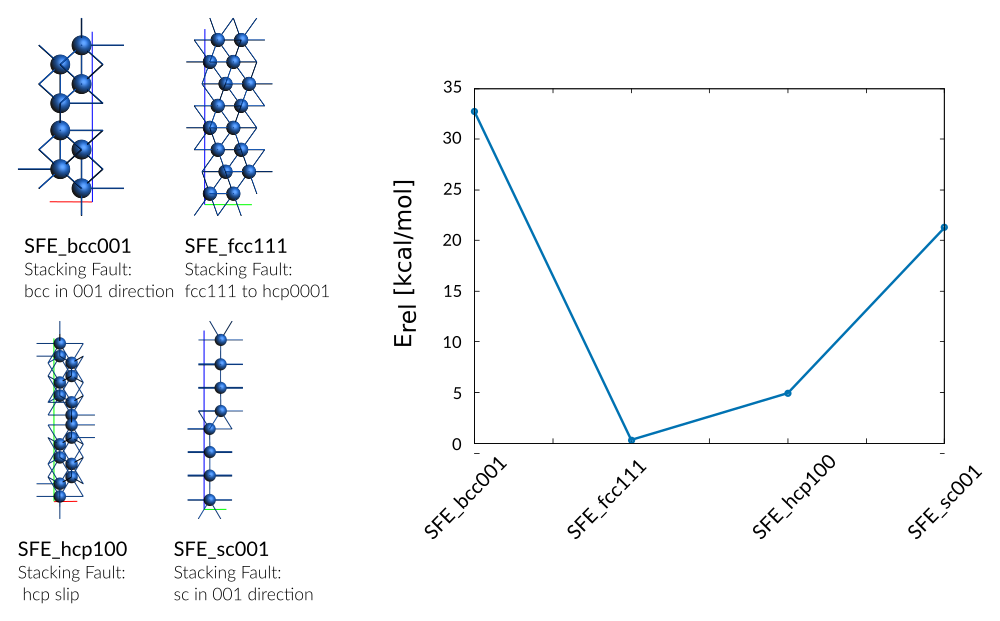
Weight Sigma Reference_value Expression
0.394 1.255 32.720 kcal/mol +energy("SFE_bcc001")/8-energy("hcp_opt")/2
6.300 1.255 0.320 kcal/mol +energy("SFE_fcc111")/16-energy("hcp_opt")/2
6.300 1.255 4.940 kcal/mol +energy("SFE_hcp100")/16-energy("hcp_opt")/2
0.394 1.255 21.280 kcal/mol +energy("SFE_sc001")/8-energy("hcp_opt")/2
3.4.7. Elastic strain moduli¶
As discussed in the paper elastic strain moduli are calculated by manipulating the lattice vectors describing the positions of the atoms. The following elastic constants c11, c12, and c44 for bulk Co phases are included in the training data:
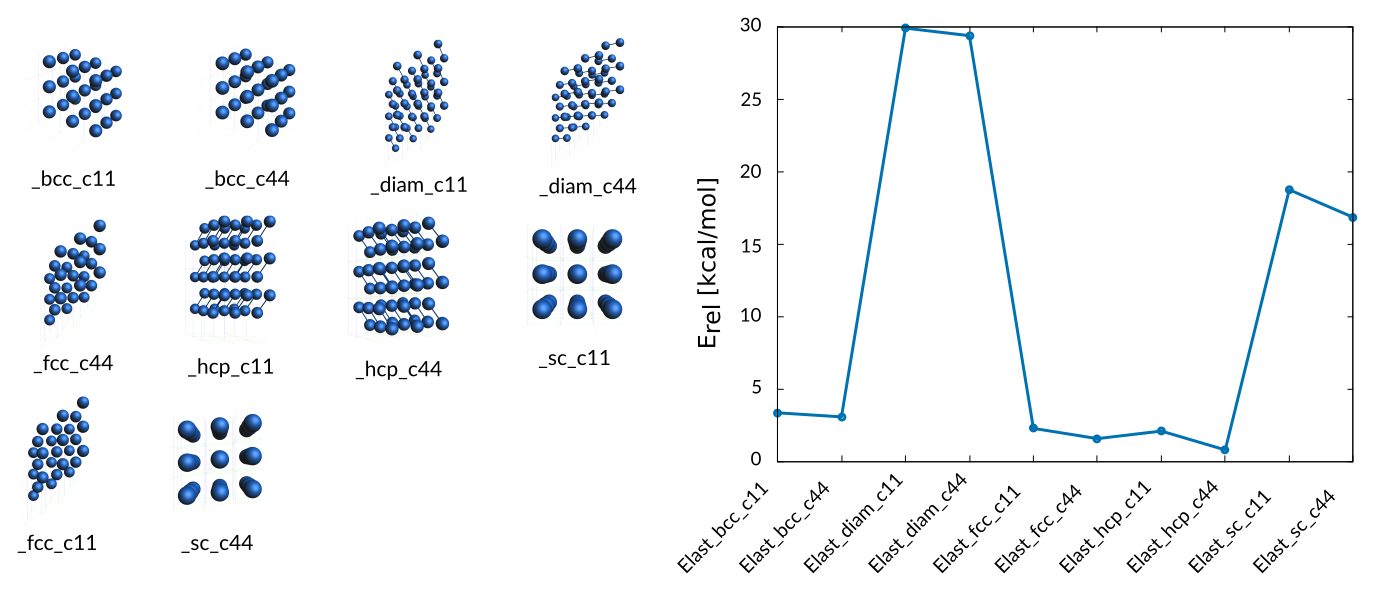
Weight Sigma Reference_value Expression
25.201 1.255 3.340 kcal/mol +energy("Elast_bcc_c11")/1-energy("hcp_opt")/2
25.201 1.255 3.060 kcal/mol +energy("Elast_bcc_c44")/1-energy("hcp_opt")/2
0.063 1.255 29.890 kcal/mol +energy("Elast_diam_c11")/2-energy("hcp_opt")/2
0.063 1.255 29.370 kcal/mol +energy("Elast_diam_c44")/2-energy("hcp_opt")/2
25.201 1.255 2.270 kcal/mol +energy("Elast_fcc_c11")/1-energy("hcp_opt")/2
25.201 1.255 1.560 kcal/mol +energy("Elast_fcc_c44")/1-energy("hcp_opt")/2
25.201 1.255 2.080 kcal/mol +energy("Elast_hcp_c11")/2-energy("hcp_opt")/2
25.201 1.255 0.780 kcal/mol +energy("Elast_hcp_c44")/2-energy("hcp_opt")/2
0.394 1.255 18.740 kcal/mol +energy("Elast_sc_c11")/1-energy("hcp_opt")/2
0.394 1.255 16.830 kcal/mol +energy("Elast_sc_c44")/1-energy("hcp_opt")/2
Tip
When manipulating the lattice in the graphical user interface (Model → Lattice) with the aim of inducing a strain, make sure you check the box Adjust atoms when changing lattice vectors.