3.1. ReaxFF (basic): H₂O bond scan¶
This example shows how to fit a ReaxFF force field to reproduce a DFT-calculated H–OH bond dissociation curve of a water molecule in the gas phase.
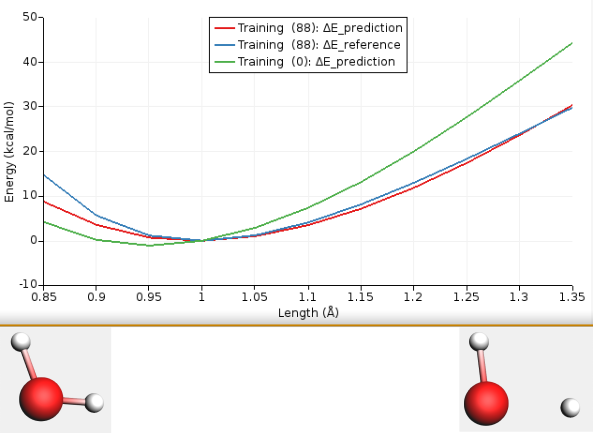
Fig. 3.1 Bond length scan of a water molecule using Water2017.ff (initial, green), DFT (reference, blue), and refitted ReaxFF force field in this tutorial (red). The (88) means that the best fit was obtained for evaluation 88.¶
The ReaxFF force field Water2017.ff (DOI: 10.1021/acs.jpcb/7b02548) is used as a starting point for the parametrization.
Note
The Water2017.ff force field was originally optimized for liquid water, not gaseous water.
The tutorial files can be found in
$AMSHOME/scripting/scm/params/examples/ReaxFF_water. The input files
already exist if you want to skip generating them (then jump to Run the ReaxFF parametrization).
For each step, a few different ways are shown:
GUI: ParAMS and AMS graphical user interface
Python: This uses PLAMS for the
reference job, and the ParAMSJob and ParAMSResults Python classes torun the optimization and generate plots.Command-line. This uses the ParAMS Main Script to run the optimization and generate plots.
The differences are summarized at the end of the tutorial.
3.1.1. Calculate the reference data¶
The reference data will be a bond scan of one of the H–OH bonds in gaseous water. The easiest way to set up such a calculation is as follows:
O(1) H(2) distance110.85 angstrom, and the maximum to 1.35 angstrom.H2O_pesscan and run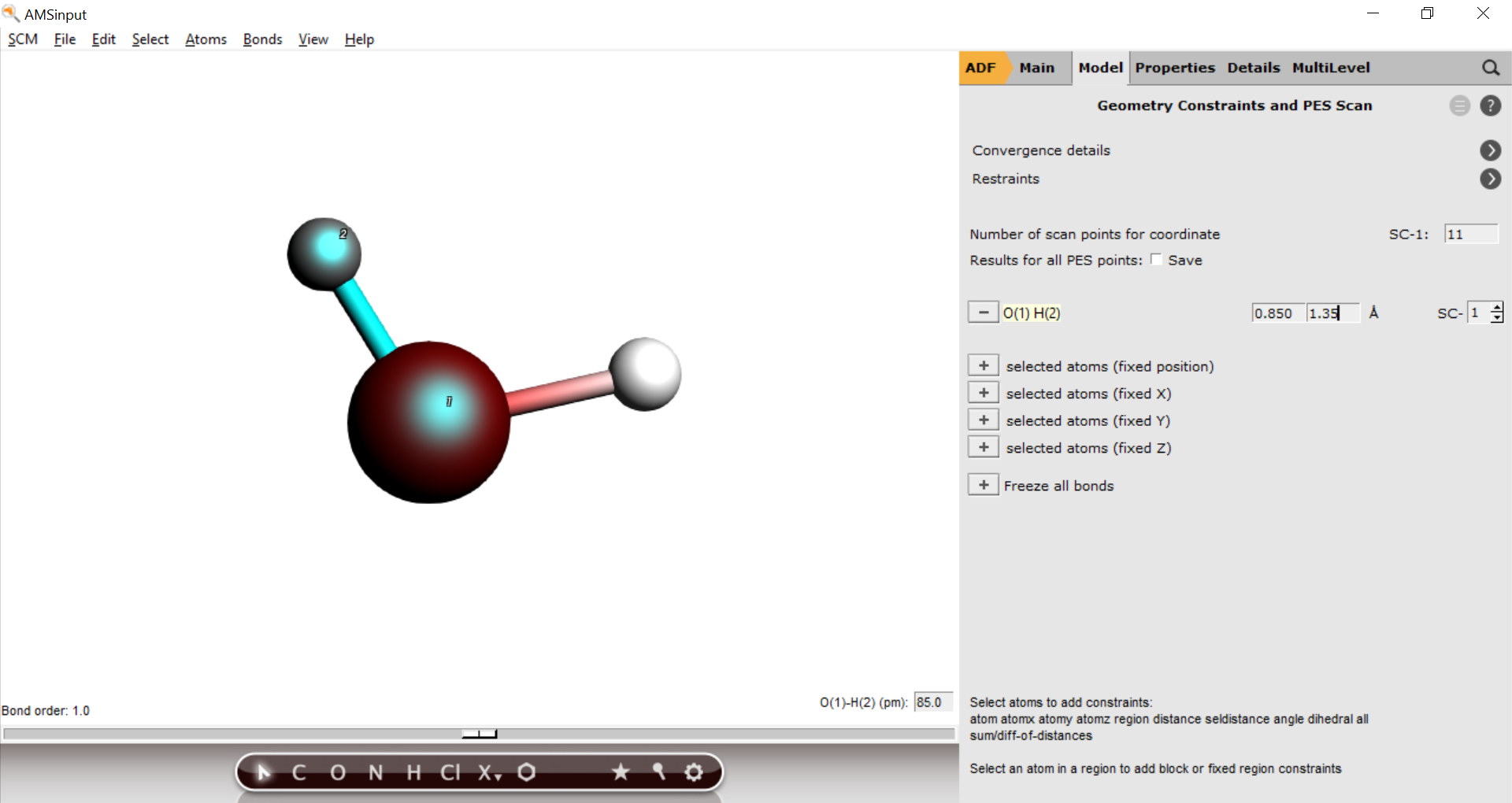
def run_reference():
# choose a reference engine. If you do not have a license for ADF, use DFTB, ForceField, or another engine for which you have the license.
# from_smiles('O') creates an H2O molecule
job = AMSJob.from_input(
name="H2O_pesscan",
molecule=from_smiles("O"),
text_input="""
Task PESScan
PESScan
# Scan O-H bond length between 0.85 and 1.35 angstrom (11 points)
ScanCoordinate
nPoints 11
Distance 1 2 0.85 1.35
End
End
Engine ADF
XC
GGA PBE
End
EndEngine
#Engine ForceField
#EndEngine
#Engine DFTB
#EndEngine
""",
)
job.run()
return job
3.1.2. Import the reference data into ParAMS¶
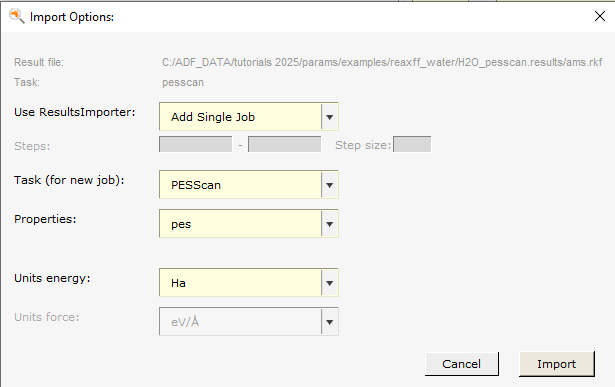
This adds
A job with the ID
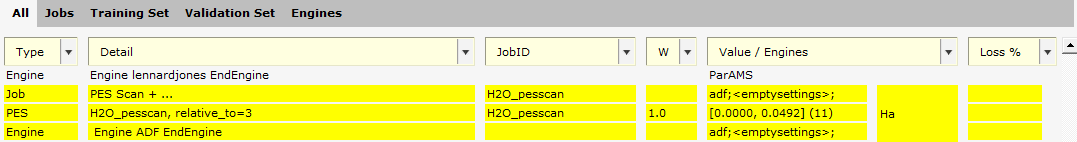
H2O_pesscanto the Job Collection. It has task PESScan, with settings taken from the reference pes scan job (which bond to scan, the number of points to sample, etc.).A training set of type PES with the detail
H2O_pesscan, relative_to=3. The relative_to=3 means that the energies along the bond scan are calculated with respect to the 4th point (the indexing starts with 0), because that point was the lowest in energy. Select the entry and switch to the Info panel at the bottom to see the reference values.An engine
adf;<empty Settings>;. This engine contains the settings that were used in the reference calculation.
If you would like to change the point along the bond scan that the other energies are relative to, it can be done as follows:
H2O_pesscan, relative_to=3. This brings up a dialog with more details.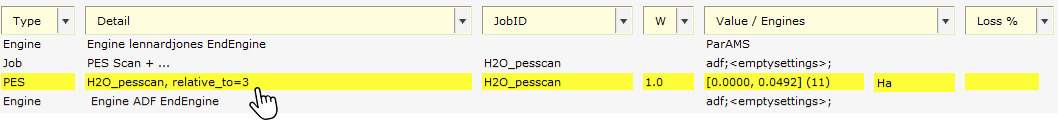
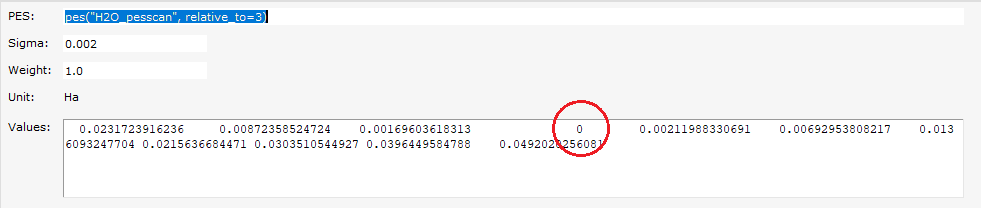
relative_to=3 to relative_to=0.
H2O_pesscan, relative_to=0. This brings up a dialog with more details.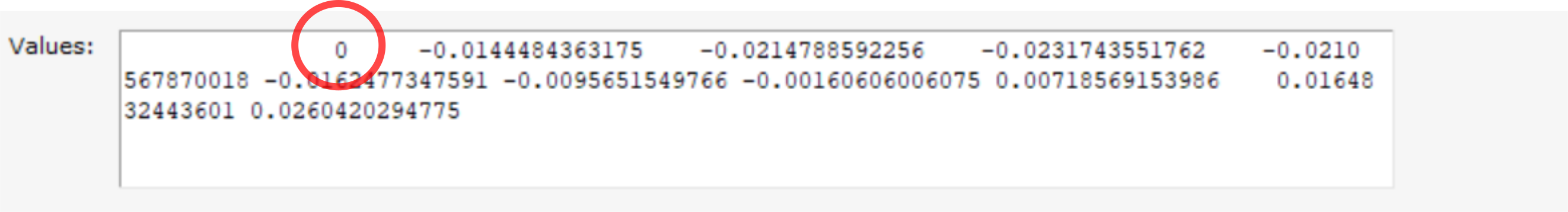
relative_to=0 to relative_to=3.See also
From generate_input_files.py.
The following lines set up a Results Importer with kcal/mol energy units, import the results, and store the training_set.yaml and job_collection.yaml files. It also illustrates the use of a weights scheme, in which the weights depend on the reference values. Run the script with $AMSBIN/amspython generate_input_files.py.
def import_results():
# If you ran the PESScan job via the graphical user interface, set
# pesscan_job = '/path/to/ams.rkf'
# Otherwise, the below line runs the reference job
pesscan_job = run_reference()
ri = ResultsImporter(settings={"units": {"energy": "kcal/mol"}})
ri.add_singlejob(
pesscan_job,
task="PESScan",
properties={
"pes": {
# apply a weights scheme (optional).
# This sets the weight for the larger energies (very short or very long bond distance) to smaller numbers.
# stdev is the standard deviation of the Gaussian in energy units (here kcal/mol)
"weights_scheme": WeightsSchemeGaussian(normalization=11.0, stdev=20.0)
}
},
)
ri.store(backup=False, store_settings=False)
3.1.3. Set the parameters to optimize¶
In this case, we will optimize all O-H bond parameters of the ReaxFF force field. The starting point (initial parameters) come from Water2017.ff (DOI: 10.1021/acs.jpcb/7b02548).
First, load Water2017.ff:
Next, only set the O-H bond parameters to be active:


H O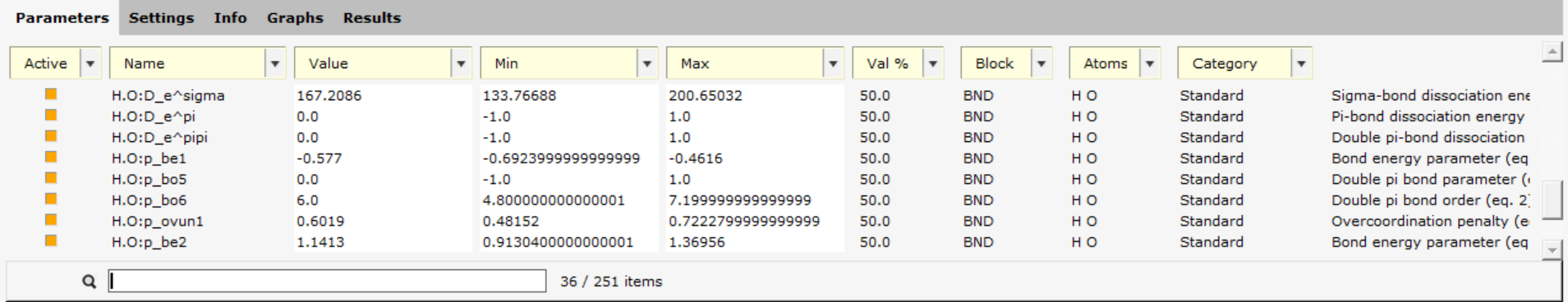
Tip
In the bottom search box, type H.O: to search for parameters
between H and O (both BND and OFD parameters). Remember to clear the
search box to see all parameters again.
Note
Normally you wouldn’t optimize all bond parameters, but only a few of them (you could try different combinations). Here, we optimize all bond parameters for illustration purposes only. For example, optimizing π-bond parameters for H₂O is not very useful!
See also
The equations that the parameters enter.
From generate_input_files.py.
The following function initializes a ReaxFF Parameter interface based on the parameters in Water2017.ff.
bounds_scale=1.2means that the minimum and maximum allowed values will be within 20% of the original value for all optimized parameters.header['head']is the first line of the output ffield.ff (force-field) fileOnly the O-H bond parameters become active.
The interface is stored in the file
parameter_interface.yaml.
def create_parameter_interface():
# start from the Water2017.ff force field
parameter_interface = ReaxFFParameters("Water2017.ff", bounds_scale=1.2)
parameter_interface.header["head"] = "Reparametrization of Water2017.ff"
# only activate the "Standard" bond parameters for O-H
for p in parameter_interface:
p.is_active = (
(p.metadata["block"] == "BND")
and (p.metadata["category"] == "Standard")
and (p.metadata["atoms"] == ["H", "O"] or p.metadata["atoms"] == ["O", "H"])
)
parameter_interface.yaml_store("parameter_interface.yaml")
3.1.4. Run the ReaxFF parametrization¶
For ReaxFF, we recommend to use CMA-ES. It is a method which samples popsize parameters around a central point (mean). The first central point is the initial parameters. The sigma keyword affects how broad the sampled distribution is (how far away from the central point parameters are sampled).
After an iteration, which consists of popsize parameter evaluations, the central point and sigma are updated. The central point will approach the optimized parameters, and sigma will decrease as the optimization progresses. In this way, the sampled distribution is very broad in the beginning of the optimization, but becomes smaller with time.
When sigma reaches the minsigma value, the optimization stops.
See also
Video tutorial on YouTube about CMA-ES with ReaxFF (AMS2021 version)
Tip
Decrease the initial sigma value for faster convergence (less global optimization).
 panel in the bottom table
panel in the bottom table100
 next to Optimizers
next to Optimizers0.1100.0001
55.
reaxff_water.paramsThe params.in file contains:
Task Optimization
DataSet
Name training_set
Path training_set.yaml
End
LoggingInterval
General 5
Parameters 5
History 5
Flush 5
End
ExitCondition
Type MaxTotalFunctionCalls
MaxTotalFunctionCalls 100
End
Optimizer
Type CMAES
CMAES
Sigma0 0.1
Popsize 10
MinSigma 0.0001
Sampler full
Verbose True
End
End
The ExitCondition block of type
MaxTotalFunctionCallsmeans that the optimization will stop after 100 evaluations.The DataSet LoggingInterval block specifies that logging will happen every 5 evaluations or whenever the loss function decreases
To run the optimization:
"$AMSBIN"/params
From optimization.py.
#!/usr/bin/env amspython
# Usage: "$AMSBIN/amspython" optimization.py
from scm.plams import *
from scm.params import *
import os
import matplotlib.pyplot as plt
def main():
init() # initialize PLAMS workdir
examples_dir = os.path.expandvars("$AMSHOME/scripting/scm/params/examples/ReaxFF_water")
# load the optimization job from examples directory and run
optimization_job = ParAMSJob.from_inputfile(examples_dir + "/params.in", name="optimization")
optimization_job.run()
# or load a finished previously run job:
# optimization_job = ParAMSJob.load_external('plams_workdir/optimization/results')
print("Optimized force field stored in " + optimization_job.results.get_ffield())
# loop over all PES scans and show plots
# in this case there is only 1 PES Scan
all_pesscans = optimization_job.results.get_all_pes_prediction_names()
for scan in all_pesscans:
x_headers, x_values, ref_values, pred_values, unit = optimization_job.results.get_pes_prediction(scan)
plt.plot(x_values[0], ref_values, "-") # ref. vs distance
plt.plot(x_values[0], pred_values, "-") # pred. vs distance
plt.legend(["Reference", "Prediction"])
plt.xlabel(x_headers[0])
plt.ylabel(f"Relative energy ({unit})")
plt.title(scan)
plt.savefig(f"plot_{scan}_from_python.png")
plt.show()
finish()
if __name__ == "__main__":
main()
See also
3.1.5. Visualize the results¶
For more details about graphs and results, see the Getting Started: Lennard-Jones tutorial.
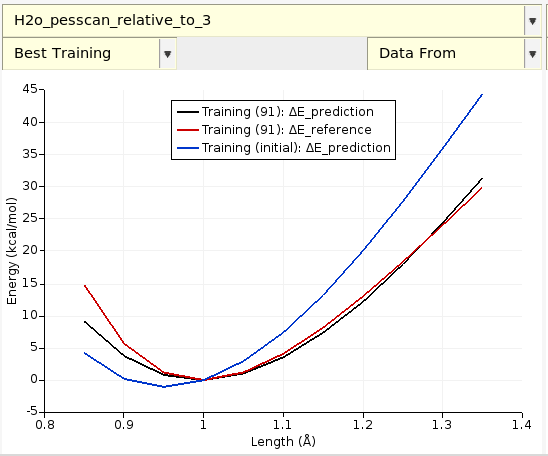
On the Graphs tab, select Best H2O_pesscan_relative_to_3.
The data in this graph are taken from optimization/training_set_results/best/pes_predictions/H2O_pesscan_relative_to_3.txt in the job results folder.
To compare to the bond scan using Water2017.ff, check Data From → Training(initial): ΔE_prediction.
Tip
You double-click on a plot axis to access plot configuration settings.
In the directory
optimization/training_set_results/best/pes_predictions, there is a file
H2O_pesscan_relative_to_3.txt. The relative_to_3 means that the energies
along the bond scan are calculated with respect to the 4th point (the indexing
starts with 0). Use params plot H2O_pesscan_relative_to_3.txt to generate
an image like the following:
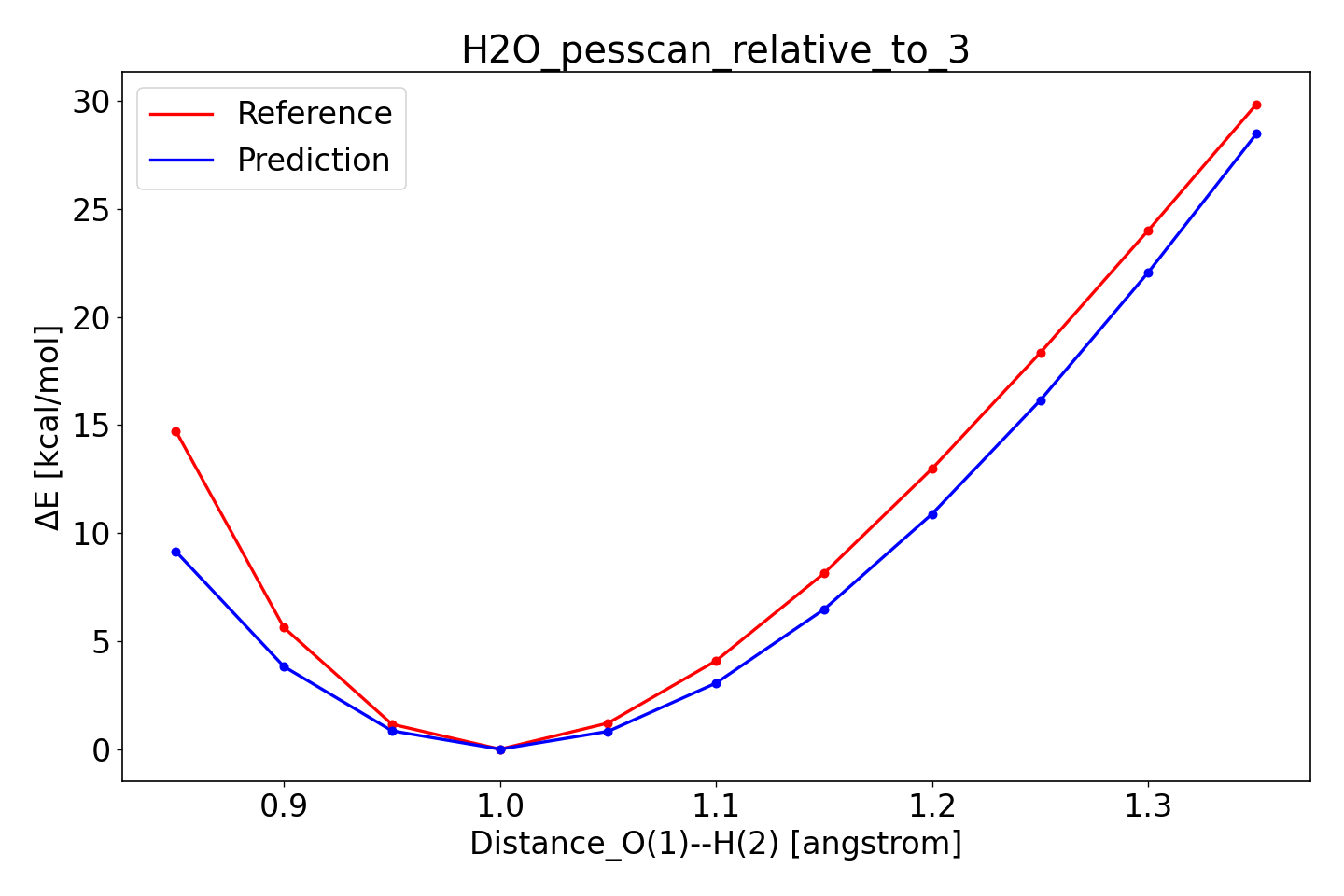
To compare to the initial parameters, make the corresponding plot from
the optimization/training_set_results/initial/pes_predictions
directory.
From optimization.py.
#!/usr/bin/env amspython
# Usage: "$AMSBIN/amspython" optimization.py
from scm.plams import *
from scm.params import *
import os
import matplotlib.pyplot as plt
def main():
init() # initialize PLAMS workdir
examples_dir = os.path.expandvars("$AMSHOME/scripting/scm/params/examples/ReaxFF_water")
# load the optimization job from examples directory and run
optimization_job = ParAMSJob.from_inputfile(examples_dir + "/params.in", name="optimization")
optimization_job.run()
# or load a finished previously run job:
# optimization_job = ParAMSJob.load_external('plams_workdir/optimization/results')
print("Optimized force field stored in " + optimization_job.results.get_ffield())
# loop over all PES scans and show plots
# in this case there is only 1 PES Scan
all_pesscans = optimization_job.results.get_all_pes_prediction_names()
for scan in all_pesscans:
x_headers, x_values, ref_values, pred_values, unit = optimization_job.results.get_pes_prediction(scan)
plt.plot(x_values[0], ref_values, "-") # ref. vs distance
plt.plot(x_values[0], pred_values, "-") # pred. vs distance
plt.legend(["Reference", "Prediction"])
plt.xlabel(x_headers[0])
plt.ylabel(f"Relative energy ({unit})")
plt.title(scan)
plt.savefig(f"plot_{scan}_from_python.png")
plt.show()
finish()
if __name__ == "__main__":
main()
This creates an image like the following:
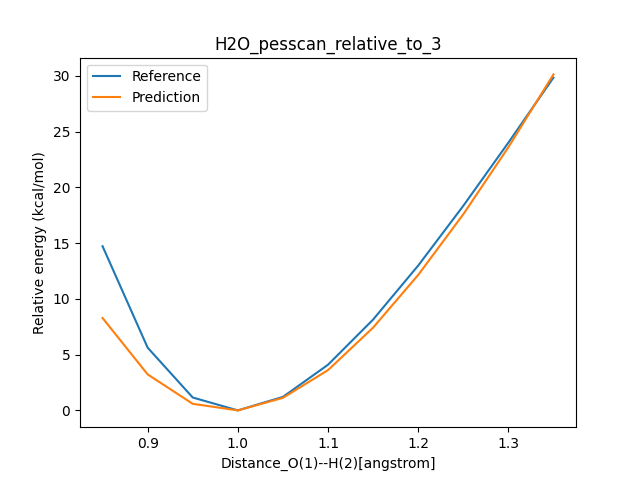
3.1.6. ffield.ff: The force field file¶
To run new ReaxFF simulations with your fitted force field, you need the force field file ffield.ff.
You can find it in the results directory in optimization/training_set_results/best/ffield.ff.
After the parametrization job has finished, in the ParAMS GUI you can select File → Open Optimized Engine in AMSinput → Best training to open AMSinput with the engine settings (path to the trained force field file) already inserted into AMSinput.
Tip
The first line of the file is a header. Fill it with some meaningful content describing
who made it,
when it was made, and
the types of training data (systems, reference methods)
You can also change the header in the ParAMS GUI: Select Parameters → Edit ReaxFF header before running your job.
3.1.7. Summary of the 3 different ways¶
Differences of the three different ways of running this tutorial (GUI, Python, Command-line) are given below:
Action |
GUI |
Python |
Command-line |
|---|---|---|---|
Run reference job |
AMSinput/AMSjobs |
PLAMS |
– |
Import results |
– |
||
Run ParAMS |
AMSjobs |
|
|
Final plot |
Graphs panel |
|
3.1.8. Next steps with ReaxFF parametrization¶
Check out the ReaxFF (advanced): ZnS for a more realistic parametrization, which also shows how to build a new ReaxFF force field from scratch.