Generating multiple conformers for use with COSMO-RS¶
The ADFCOSMORSConfJob is a customizable class that allows the user to design a conformer generation workflow for COSMO-RS. The default instance of this class generates a set of conformers and then performs ADF and subsequent COSMO calculations (equivalent to the AMS Task:COSMO-RS Compound) to generate .coskf files for all unique conformers. This class can be customized by adding both filters (to limit the number of conformers) and additional calculation steps (to improve the final geometry given to ADF and/or to increase the accuracy of energy calculations for filters). The generated .coskf can subsequently be utilized by the multi-species COSMO-RS, which accounts for compounds existing in multiple conformations. The application of multi-species COSMO-RS can be explored through tutorials available in both the GUI and python scripting with pyCRS.
Example usage¶
First, we’ll import the necessary classes:
from scm.plams.recipes.adfcosmorsconformers import ADFCOSMORSConfJob, ADFCOSMORSConfFilter
from scm.plams import Molecule, from_smiles, init, finish, Settings
from scm.conformers import ConformersJob
Now, we’ll input the acetic acid molecule with the from_smiles function
mol = from_smiles("CC(=O)O")
Now, we’ll specify a conformer generator (identical to the default) that generates only 50 initial structures:
conf_sett = Settings()
conf_sett.input.AMS.Generator.RDKit
conf_sett.input.AMS.Generator.RDKit.InitialNConformers = 50
conf_job = ConformersJob(name="conformers_uff", molecule=mol, settings=conf_sett)
Let’s also specify an additional step to add to the default workflow. Here, we’ll add a DFTB geometry optimization.
dftb_sett = Settings()
dftb_sett.input.AMS.Task = "Optimize"
dftb_sett.input.DFTB
The final thing we need to specify are filters. Let’s make three filters, the first to take a maximum of 20 conformers with a maximum energy range of 22 kcal/mol, the second with 10 conformers and 12 kcal/mol and the third with 5 conformers and 7 kcal/mol.
#ADFCOSMORSConfFilter(max number of conformers, max energy range)
fil1 = ADFCOSMORSConfFilter(20,22) # applied to UFF
fil2 = ADFCOSMORSConfFilter(10,12) # applied to DFTB
fil3 = ADFCOSMORSConfFilter(5,7) # applied to ADF gas phase
Finally, we give this information to the ADFCOSMORSConfJob class. We also specify the name of the coskf files as well as the directory in which we’ll find them after the calculations complete.
a = ADFCOSMORSConfJob(
mol,
conf_gen = conf_job,
first_filter = fil1,
additional = [(dftb_sett,fil2)],
final_filter = fil3,
coskf_name = "acetic_acid",
coskf_dir = "test_coskfs"
)
a.run()
In the test_coskfs directory, we find two .coskf files with the following geometries:
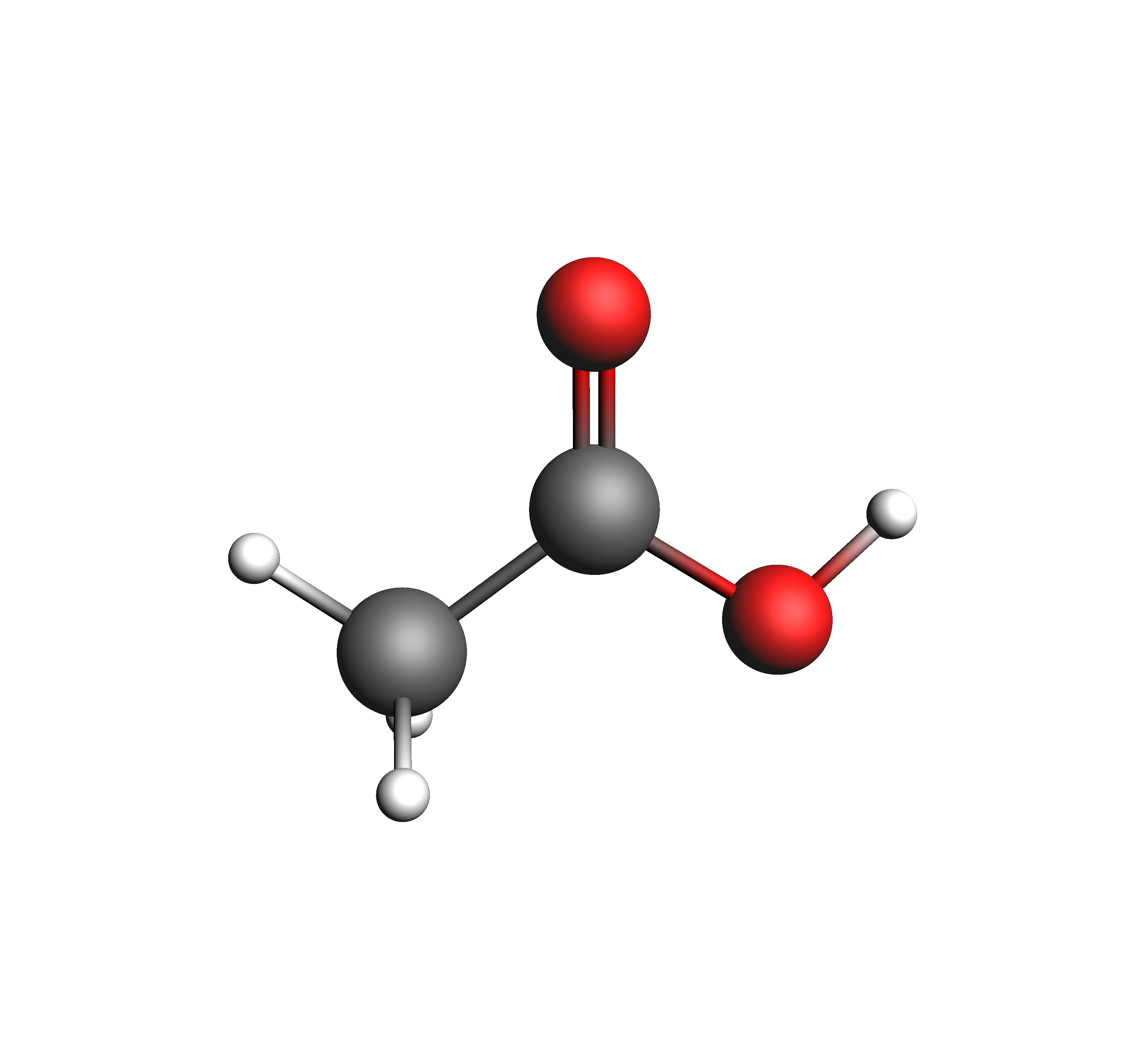
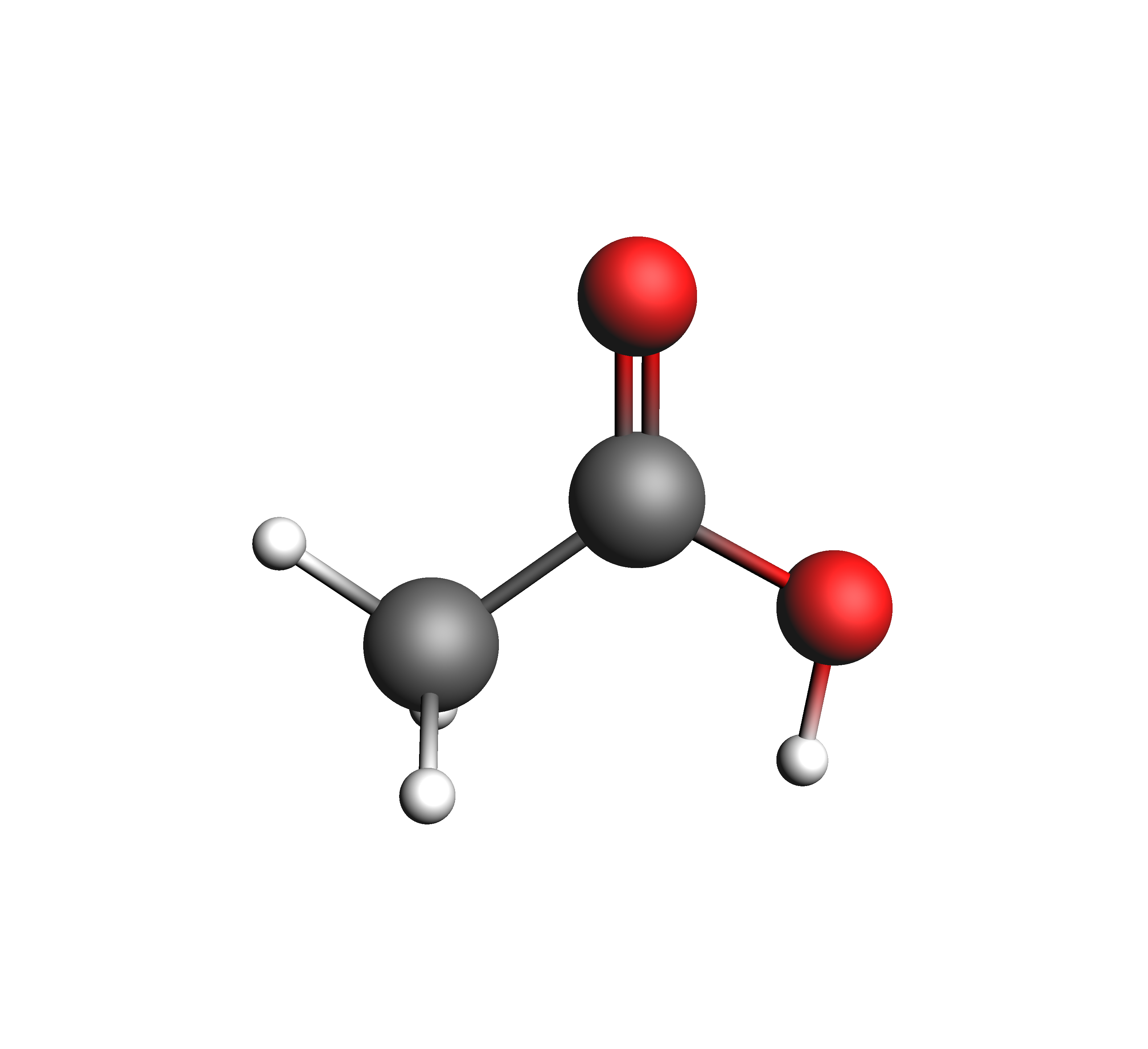
The entire script can be seen/copied when expanded below.
[show/hide code]
from scm.plams.recipes.adfcosmorsconformers import ADFCOSMORSConfJob, ADFCOSMORSConfFilter
from scm.plams import Molecule, from_smiles, init, finish, Settings
from scm.conformers import ConformersJob
init()
mol = from_smiles("CC(=O)O")
conf_sett = Settings()
conf_sett.input.AMS.Generator.RDKit
conf_sett.input.AMS.Generator.RDKit.InitialNConformers = 50
conf_job = ConformersJob(name="conformers_uff", molecule=mol, settings=conf_sett)
dftb_sett = Settings()
dftb_sett.input.AMS.Task = "Optimize"
dftb_sett.input.DFTB
# ADFCOSMORSConfFilter(max number of conformers, max energy range)
fil1 = ADFCOSMORSConfFilter(20, 22) # applied to UFF
fil2 = ADFCOSMORSConfFilter(10, 12) # applied to DFTB
fil3 = ADFCOSMORSConfFilter(5, 7) # applied to ADF gas phase
a = ADFCOSMORSConfJob(
mol,
conf_gen=conf_job,
first_filter=fil1,
additional=[(dftb_sett, fil2)],
final_filter=fil3,
coskf_name="acetic_acid",
coskf_dir="test_coskfs",
)
a.run()
finish()
Important Information¶
Specifying Filters¶
Filters are given as instances of the ADFCOSMORSConfFilter class. This class takes two arguments concerning the number of conformers and maximum energy range. These are documented in more detail below. There are 3 places to specify filters:
in the
first_filterkeyword inADFCOSMORSConfJob. This filter will be applied to the initial set of sampled conformers.in the
final_filterkeyword inADFCOSMORSConfJob. This filter will be applied to the results of the ADF gas phase calculation.in the
additionalkeyword as the second element of a (Settings,ADFCOSMORSConfFilter) tuple. The filter will be applied on the results of the calculation done with theSettingsfrom the same tuple. To be perfectly clear, this means a (Settings,ADFCOSMORSConfFilter) tuple adds a step to the workflow where a calculation is done with theSettingsobject and then those results are immediately filtered with the correspondingADFCOSMORSConfFilter. A list of these tuples can be given to perform a series of calculations/filtering steps.
Specifying Settings for additional jobs¶
Settings objects provided to the additional keyword should be valid. ADFCOSMORSConfJob will likely fail if there are errors in the Settings objects. In the case of unexpected failures, take a look in the calculation subdirectories called additional_i where i is an index representing the index of the additional calculation.
Note
The tasks for Settings objects should correspond to Tasks for AMSConformers. The most important two types are Optimize to perform a geometry optimization and Score to perform a single point calculation. The tasks section in the Conformers doc contains all additional information.
Specifying conformer sampling strategies¶
The default conformer sampling strategy is based on RDKit. This is likely sufficient for more use cases, but the user can specify another sampling strategy by providing a ConformersJob instance to the conf_gen keyword. See also The generator section of the conformer documentation for more information about specifying alternative generators.
Brief API Documentation¶
-
class
ADFCOSMORSConfFilter(max_num_confs=None, max_energy_range=None)[source]¶ This class allows the user to specify criteria on which to filter sets of conformers.
- Parameters
max_num_confs – The maximum number of conformers to bring forward to the next step. These are automatically ordered by energy, so the first n lowest energy structures are carried carried forward to the next step.
max_energy_range – The maximum allowable difference (in kcal/mol) between the lowest energy conformer and the highest energy conformer that will be carried forward. For example, for a max_energy_range of 2 kcal/mol and a lowest energy structure of 1 kcal/mol, any structure with an energy above 3 (1+2) kcal/mol will be filtered out.
-
class
ADFCOSMORSConfJob(molecule, conf_gen=None, first_filter=None, additional=None, final_filter=None, adf_singlepoint=False, initial_conformers=500, coskf_dir=None, coskf_name=None, mol_info={}, **kwargs)[source]¶ This class allows for the user to implement a custom workflow for generating multiple conformers for use with COSMO-RS. The class allows the user to input conformer generation strategies, job types to refine the energies/geometries, and filters to remove conformers between each calculation step.
- Parameters
molecule (
Molecule) – A plams Molecule instance- Keyword Arguments
conf_gen (ConformersJob) – A ConformersJob instance
first_filter (ADFCOSMORSConfFilter) – The ADFCOSMORSConfFilter to apply to the initial set of sampled conformers
additional – A list of (
Settings, ADFCOSMORSConfFilter) tuples. The elements of this list represent additional calculation steps (e.g., progressively higher levels of theory) and possibly different filters to apply at each step.final_filter (ADFCOSMORSConfFilter) – The ADFCOSMORSConfFilter to apply to the results of the gas phase ADF calculation for the conformers
adf_singlepoint (bool) – A boolean indicating if the adf gas phase calculation in the cosmo-rs compound task should be a single point. This defaults to False.
initial_conformers – the (integer) number of initially sampled conformers. This is only applied if the default conformer generation strategy is used.
coskf_dir – a directory to put all the .coskf files generated for the conformers. If this keyword is not specified, the .coskf files are put in the directory containing the adf gas phase calculation results
coskf_name – a base name to be used with conformers. All conformers will have the name coskf_name*_i where i is the index of the unique conformer. If not specified, the base name becomes simply *conformer
name – an optional name for the calculation directory.
mol_info (dict) – an optional dictionary containing information will be written to the Compound Data section within the COSKF file.