COSMO result files¶
If you already have COSMO result files for all the compounds that you are interested in you can skip this tutorial, without problems of continuity. For example, ADF has a database of COSMO result files, the COSMO-RS compound database ADFCRS-2010.
The purpose of this tutorial is to teach you how to make data for a compound using the ADF program such that it can be read by COSMO-RS. COSMO-RS expects so called COSMO result files, which are results of quantum mechanical calculation using COSMO. In ADF such a COSMO result file is called a TAPE21 (.t21) file, or a COSKF (.coskf) file. For example the COSMO-RS compound database ADFCRS-2010 consists of .coskf files. In other programs such a file can be a .cosmo file. For example, at http://www.design.che.vt.edu/VT-Databases.html a database of .cosmo files can be found, which were made with a different program. Note that the optimal COSMO-RS parameters may depend on the program chosen.
Please read through the first GUI tutorial before starting with this tutorial. Even better: try using the ADF-GUI yourself, especially the Getting started Tutorial.
In this tutorial an ADF COSMO result file and a MOPAC COSMO result file is made. For ADF COSMO-RS calculations the recommended choice is to use ADF COSMO result files.
Step 1: Start ADFinput¶
For this tutorial we prefer to work in a separate directory, for example a directory called Tutorial, as was explained in the Getting started Tutorial.
Start ADFjobs (in your home directory), and move to the Tutorial directory:
- Start adfjobsClick on the Tutorial folder icon
Next start ADFinput using the SCM menu.
- Select the SCM → New input menu command.
Step 2: Create the molecule¶
First we construct a water molecule, and preoptimize its geometry:
- Select the O-tool by clicking on the button with the ‘O’Click somewhere in the drawing area to create an oxygen atomSelect the select-tool by clicking on the button with the arrowClick once in empty space so nothing is selectedSelect Atoms → Add HydrogenClick the optimizer button
Your water molecule should look something like this:
Step 3: ADF COSMO result file¶
The next step is to optimize the gas geometry using ADF, and perform the ADF COSMO calculation at the optimized gas phase geometry.
- Enter a proper title in the Title field (like ‘water’)Select Preset → COSMO-RS Compound
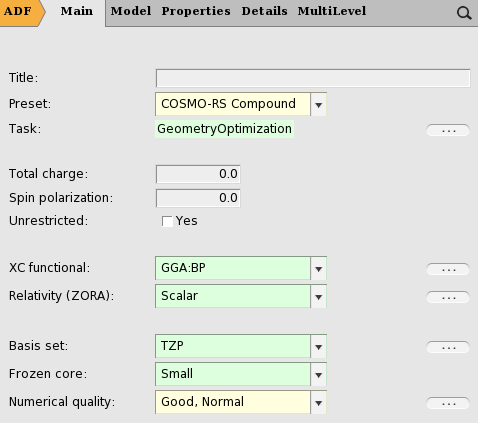
For your information, the proper settings for the gas phase geometry optimization are: the Becke Perdew exchange correlation functional (GGA:BP), use of the scalar relativistic ZORA Hamiltonian, a TZP small core basis set (for Iodine a TZ2P small core basis set), and an integration accuracy with a good quality. Like for Iodine for heavier elements than Krypton, a TZ2P small core basis set is recommended. Note that these settings were used for the optimization of the COSMO-RS parameters.
With the proper options selected, now run ADF:
- Select File → RunIn the file select box, choose a name for your file (for example ‘water’)and click ‘Save’
Now ADF will start automatically, and you can follow the calculation using the logfile that is automatically shown.
- Wait until the optimization and ADF-COSMO calculation are ready (should take very little time)Click ‘Yes’ in the pop-up to read the coordinates from a .t21 file.
Now the geometry of the water molecule is the optimized one, and the ADF COSMO calculation has been performed. The result file water.coskf, which is an ADF COSMO result file, can be used as input for a COSMO-RS calculation.
Note that a .coskf file is not a complete .t21 file. For example, if one has such a .coskf file, only the COSMO surface charge density can be viewed with ADFview. Thus a .coskf file is mostly useful for COSMO-RS calculations.
If you do not want to optimize the geometry, set 1 to the number of geometry iterations in Details → Geometry Convergence. More details on parameters used in the COSMO calculation can be found in Model → Solvation, and Details → COSMO. See also the COSMO-RS manual.
Step 4: MOPAC COSMO result file¶
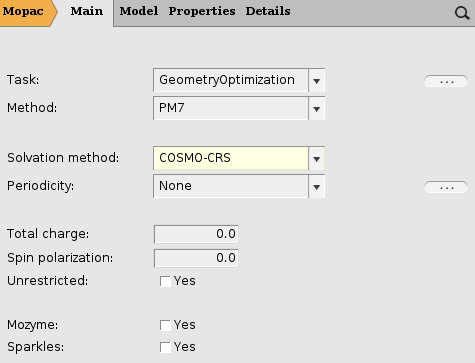
A MOPAC COSMO result file can be created in almost the same way as an ADF COSMO result file. In this step we will skip the first steps of creating the water molecule. We will change the program from ADF to MOPAC, and select the COSMO solvation method.
- Select ADF → MopacSelect Solvation Method → COSMO-CRS
For sake of clarity we will save the COSMO calculation under a different name, and run the calculation
- Select the ‘Save As...’ command from the ‘File’ menuIn the file select box, choose ‘water_mopac’ as name for your file and click ‘Save’Select File → RunWait until the optimization is ready (should take very little time)Click ‘Yes’ in the pop-up to read the coordinates from a .rkf file.
After the calculation has finished the file water_mopac.coskf, which is a MOPAC COSMO result file, can be used as input for a COSMO-RS calculation.
Note that MOPAC is a semi-empirical quantum chemistry program, whereas ADF is based on density functional theory (DFT). Thus the MOPAC COSMO result files will not be of the same quality as the ADF COSMO result files.
Part of the COSMO parameters that are set can be found if one selects Details → Run Script. Technically ADFinput will add some keywords to the input for MOPAC (see also http://www.openmopac.net) in case of a MOPAC COSMO calculation:
- PM7 EPS=9999.9 RSOLV=1.3 COSWRT NSPA=362