Proton affinities with third order DFTB (DFTB3)¶
In this tutorial we will calculate a proton affinity, defined as the negative gas phase enthalpy for the protonation reaction A- + H+ → AH. Gaus, Cui and Elstner have shown that third-order terms (DFTB3) generally improve the proton affinities with respect to second order self-consistent charges (SCC).
We will calculate the proton affinity of the acetate anion, CH3 COO- . In the first step of this tutorial, we will optimize acetic acid at the DFTB3 level. The second step will perform the computation on the anionic species, and compute its proton affinity.
Contact us to enable parameter files from DFTB.org, including the ThirdOrder set for DFTB3, needed to run this tutorial. For non-profit users there is no additional charge for this.
Step 1: Optimization of the neutral molecule¶
- Start ADFjobsSCM → New InputSwitch to DFTB mode (panel bar ADF → DFTB)
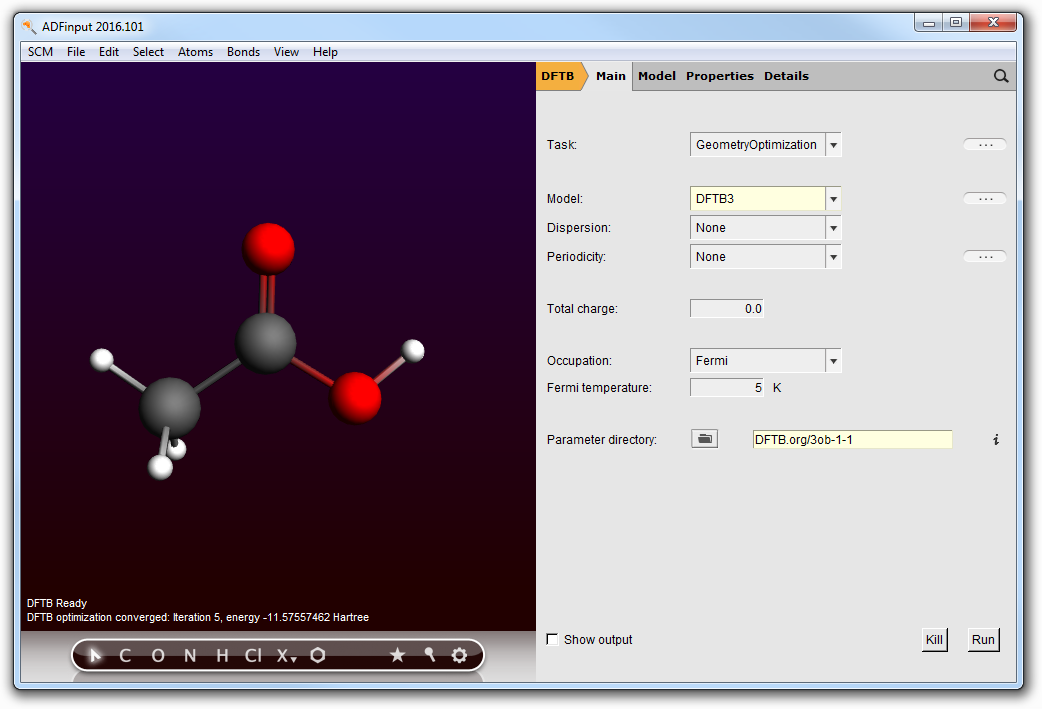
- Use the Structure tool, select Solvents → Acetic acidClick anywhere in the visualization pane. The acetic acid molecule will appear.Make sure the GeometryOptimization task is selected and charge is set to 0.0Select DFTB3 as the model.Select the ThirdOrder parameter set.Click ‘Run’ in the DFTB panel to optimize the molecule.
When the optimization is complete (DFTB Ready in the lower left corner of the window), note the final energy (for the parameter set used in this example, E(HAc) = -11.57557462 Hartree)
Step 2: Optimization of the acetate and the hydrogen ions¶
In order to perform the calculation on the acetate ion, we will remove the hydrogen ion from the previously computed acetic acid molecule.
- Select the hydrogen in the COOH group by clicking on it.Press the Backspace key on your keyboard. This will delete the hydrogen atom.Select the Main panelSet the charge to -1.0Click ‘Run’ in the DFTB panel to optimize the molecule.
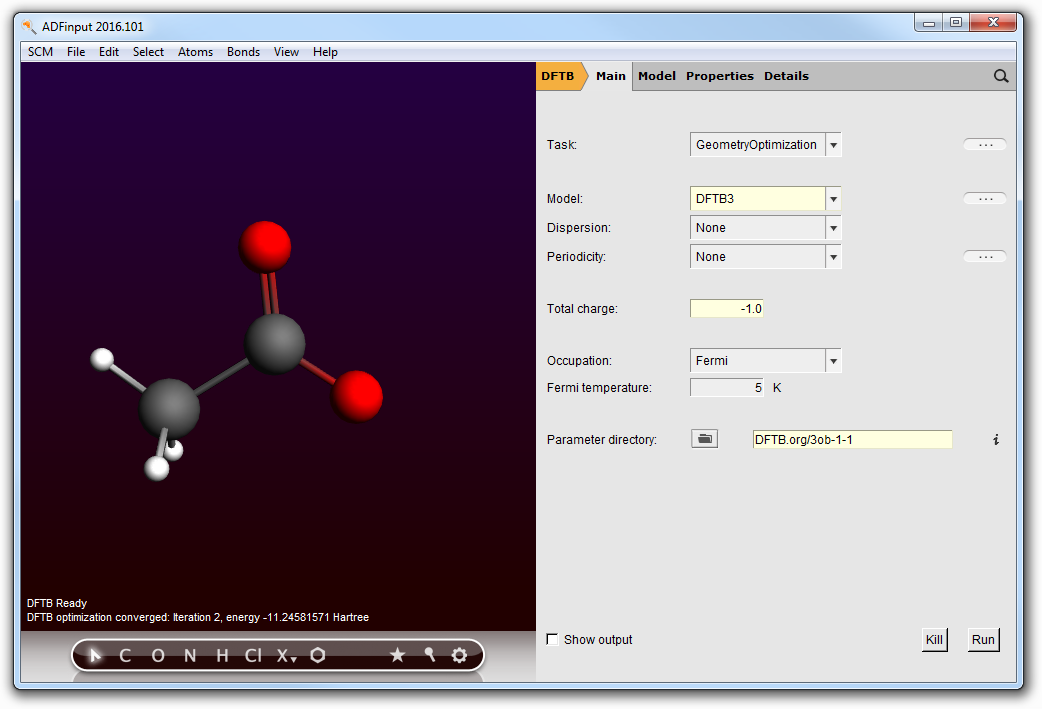
At the end of the procedure, note the final energy (for the parameter set used in this example, E(Ac- ) = -11.280518 Hartree).
Additionally, the energy of the lone proton must be obtained.
- Select and delete every atom of the molecule, except one hydrogen atom.Select the Main panelMake sure the GeometryOptimization task is selected. Although this is effectively a single point calculation, the GeometryOptimization task reports the energy directly in the molecule window, facilitating the read out of the energy.Set the charge to 1.0Click ‘Run’ in the DFTB panel.
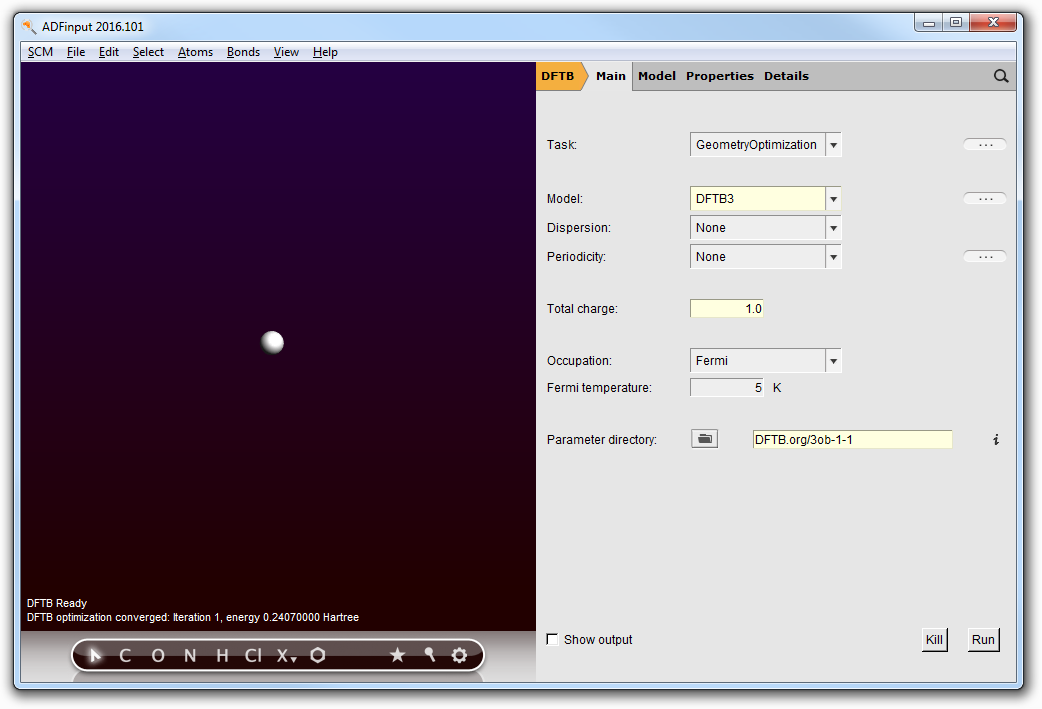
Finally, annotate the value for E(H+ ) = 0.236417 Hartree. The proton affinity is computed as PA = E(Ac- ) + E(H+ ) - E(HAc), resulting in a final Proton Affinity of 0.566798 Hartree, or 355.67 kcal/mol.
We leave it as an example to calculate the PA with DFTB2 (SCC), and compare it with the high-level ab initio results that are also quoted in the DFTB3 paper.