Overview: parameters and analysis¶
Step 1: Start AMScrs¶
For this tutorial it is convenient to start with an empty directory, for example, with the name Tutorial. You should start AMSjobs and move to the Tutorial directory:
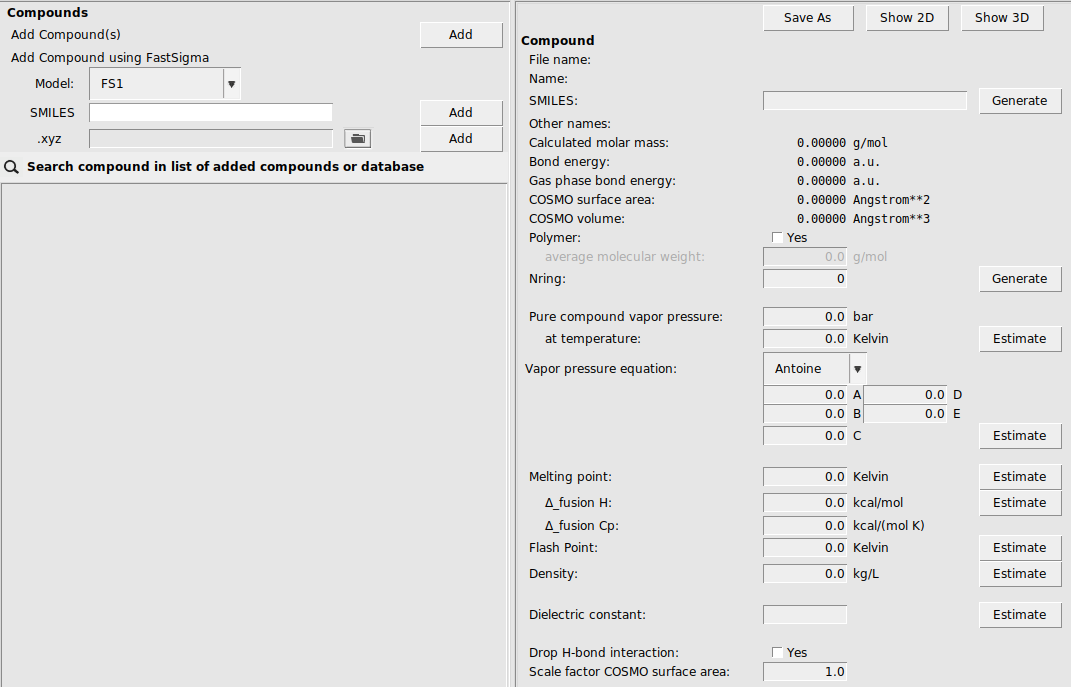
This AMScrs window consists of the following main parts:
the menu bar with the menu commands
on the left: a list of compounds and the possibility to add compounds
on the right: some properties for the selected compound
This is the so called List of Added Compounds, which is the window you will get if you select Compounds → List of Added Compounds.
If you run the COSMO-RS GUI the first time, you will be asked if you would want to download and install the COSMO-RS compound database:
The download of such databases is handled by AMSpackages. It is recommended to download the COSMO-RS compound database. On the other hand, for this tutorial, it is not necessary that the COSMO-RS compound database is downloaded.
Step 2: Add Compounds¶
In tutorial 1 it was shown how to make ADF COSMO result files. In this tutorial we will use some pre-made ADF COSMO result files.
These files can be found in the directory $AMSHOME/examples/COSMO-RS/Parameters_and_Analysis.
Copy these COSMO result files (water.coskf, methanol.coskf, ethanol.coskf, and benzene.coskf) to the directory Tutorial.
As the names suggest, these are COSMO result files of water, methanol, ethanol, and benzene, respectively.
Note that these COSKF (.coskf) files contain only the part of an ordinary TAPE21 (.t21) file which is needed in a COSMO-RS calculation. These COSKF files can only partly be used in AMSview, for example.

A file select box will open, that looks like (may look different on different platforms)
or

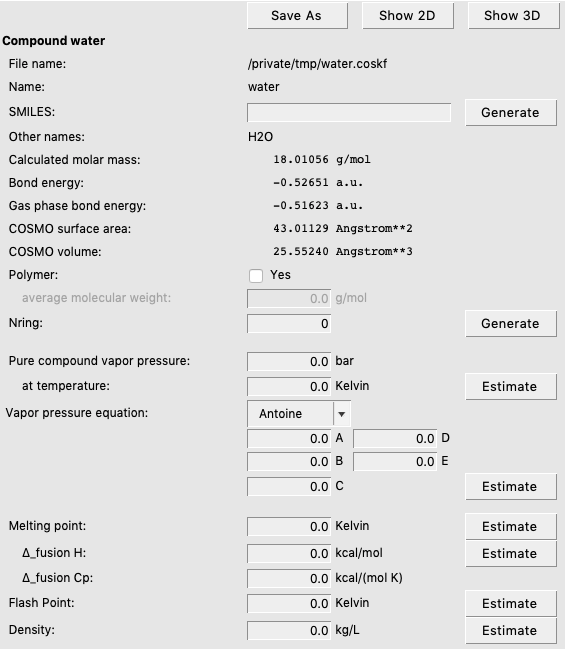
On the right side of the List of Added Compounds you can find some data that was written from the last file that was opened, in this case water.coskf. It is also possible to add some pure compound input data here, although this is not required in this particlar case. However, for some other types of compounds, user input is required at this point. We will encounter an example of this in the next step.
Step 3: Set pure compound parameters¶
In the COSMO-RS model [1] there is a ring correction term. This is important for, for example, the benzene molecule, which has 6 ring atoms. However, it is really required only when the vapor pressure of the compound is going to be computed (either because that is explicitly requested or because it is used in predicting partial vapor pressures in a mixture or gas/liquid partitioning coefficients).
For some properties, like solubility of a solid, one can or one has to include some pure compound properties in the left window of the List of Added Compounds for a selected compound.
It is possible to save this pure compound data in as a new COSKF file (or COSMO file).
To save this data in a .coskf file:
If you have followed this tutorial from the start then you will overwrite an existing benzene.coskf file. You will be asked if you want to to that, click ‘Yes’. The writing can take some time, especially for larger compounds. During this time the COSMO-RS GUI is not available for other actions.
Notice that there are ‘Estimate’ buttons to estimate some pure compound properties. The method that is used for the estimation is a group contribution method. If you press all ‘Generate’ and ‘Estimate’ buttons, you will see something like the following for benzene:
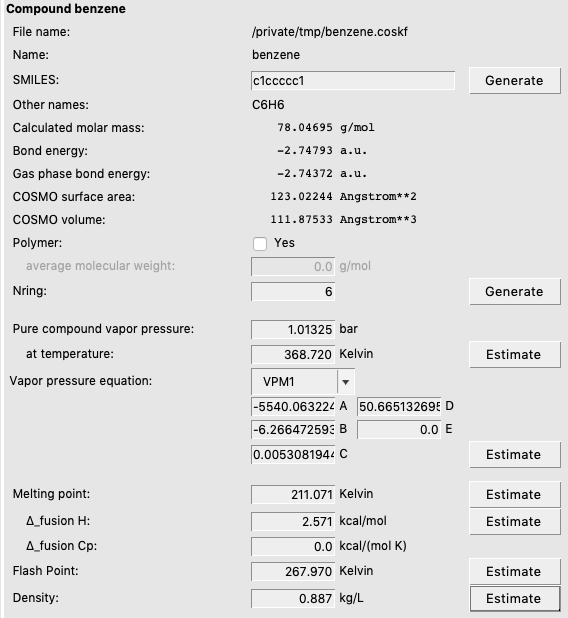
Note that some of the estimates are less accurate, such as the estimated melting point of small molecules.
Step 4: COSMO-RS, COSMO-SAC, and UNIFAC parameters¶
You can easily change between the COSMO-RS, COSMO-SAC, and UNIFAC method that is going to be used in the calculation by selecting Method → COSMO-RS, Method → COSMO-SAC, or Method → UNIFAC. Here we will use COSMO-RS, since ADF was parametrized for this method.
Expert option: set COSMO-RS parameters
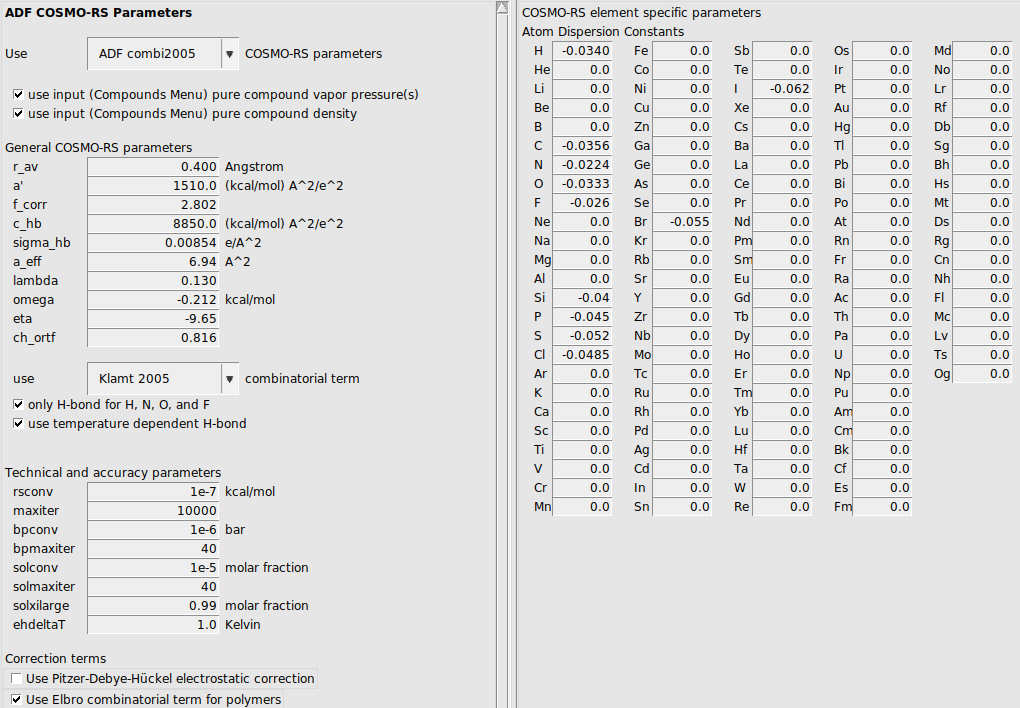
Default ‘ADF combi2005’ COSMO-RS parameters are selected, which are ADF optimized COSMO-RS parameters. See also a discussion of the COSMO-RS parameters in the COSMO-RS manual. If you select the ‘Klamt’ option for ‘Use: … COSMO-RS parameters’, the optimized parameters are chosen, which are optimized by Klamt et al., see ref. [1].
Expert option: set COSMO-SAC parameters
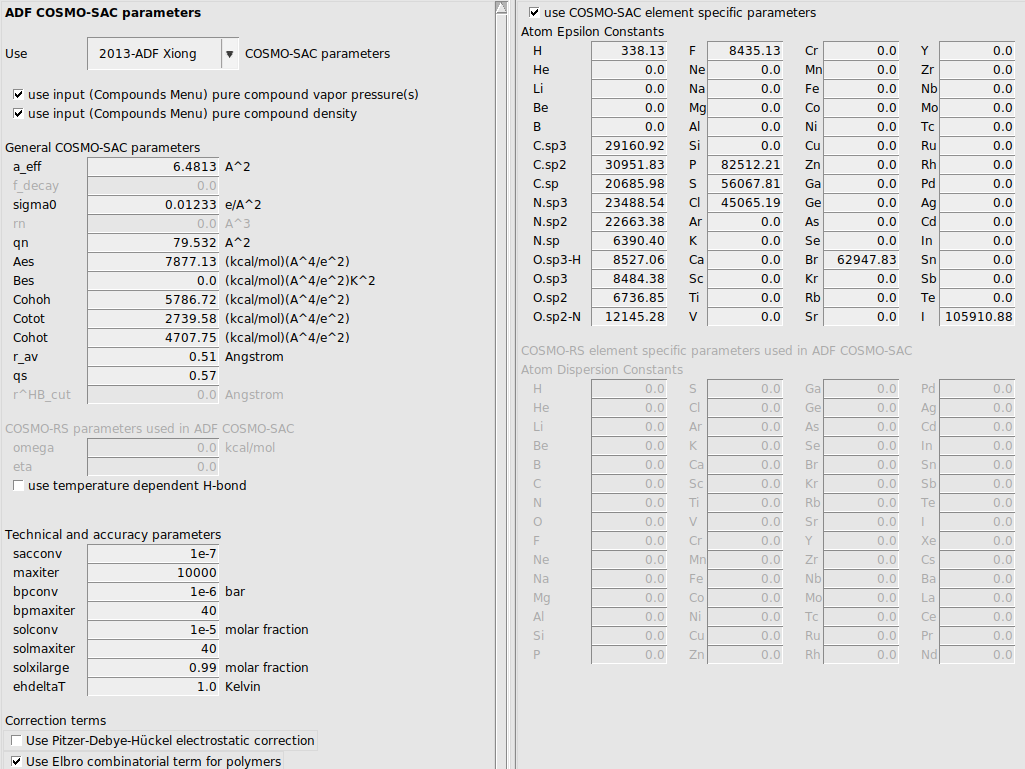
The 2013-ADF COSMO-SAC parameters were optimized for use with ADF. See also a discussion of the COSMO-SAC parameters in the COSMO-RS manual.
Expert option: set UNIFAC parameters
The UNIFAC implementation needs a SMILES string to generate the groups in a compound. The generation of SMILES strings is not done automatically.
For example, for benzene, the SMILES string ‘c1ccccc1’ should be generated (by Openbabel). You should double check the SMILES string generated, especially for charged systems, radicals, and inorganic compounds.
For the rest of the tutorial the COSMO-RS method will be used:
Step 5: Visualize the COSMO surface: AMSview¶
You can use AMSview to have a look at the COSMO surface, and the COSMO surface charge density. This is possible if the COSMO result file of the compound is a .coskf file or a .t21 file.
Note
The AMSview module was redesigned in AMS2026. If you use an older version, follow the older tutorial.
Then you will see something like:
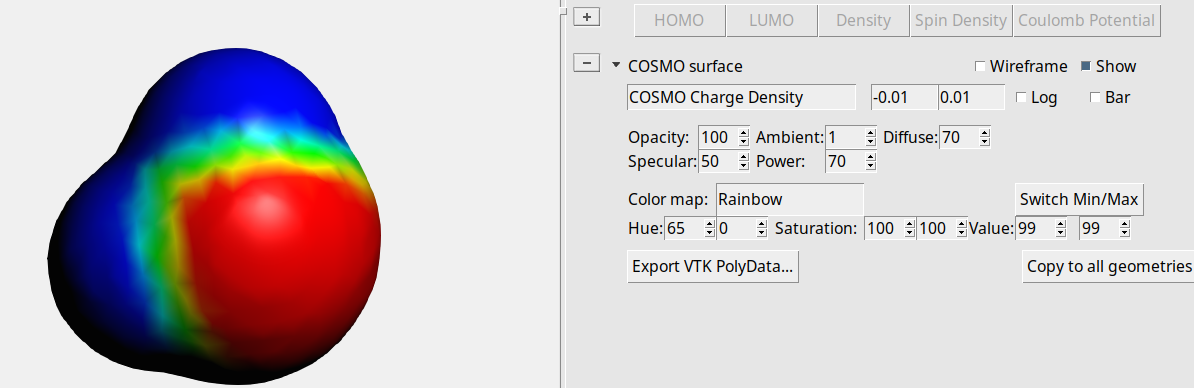
The red part represents positive COSMO charge density (the underlying molecular charge is negative), the blue part negative COSMO charge density (the underlying molecular charge is positive). You can also look at the COSMO surface points themselves.

The small spheres represent the COSMO surface points that are used for the construction of the COSMO surface.
Next we will close this AMSview window.
AMSview has many options to change the look of the picture.
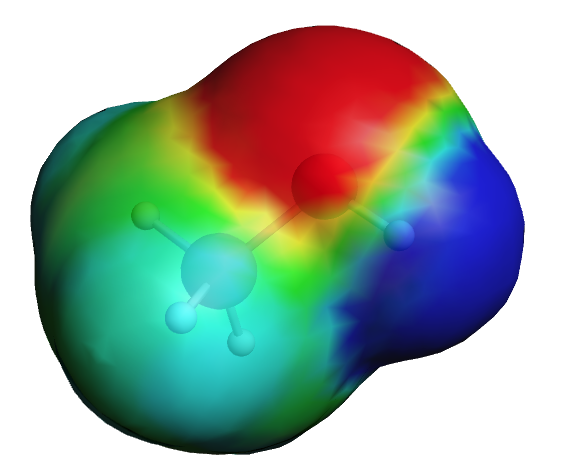
Next we will close this AMSview window.
Optionally you can change the default settings for the colormap that is used in AMSview
Step 6: Analysis: The sigma profile¶
The \(\sigma\)-profile shows the amount of surface area for a given COSMO charge density.
The sigma profiles (\(\sigma\)-profile) of the four pure compounds will be shown in a graph and in a table in the right part of the window. The whole window can be resized. The relative size of the left part of the window compared to the right part can be changed if you move the divider that is in between these parts. In the right part of the window you can also change the relative size of the upper part compared to the lower part if you move the divider that is in between these parts.
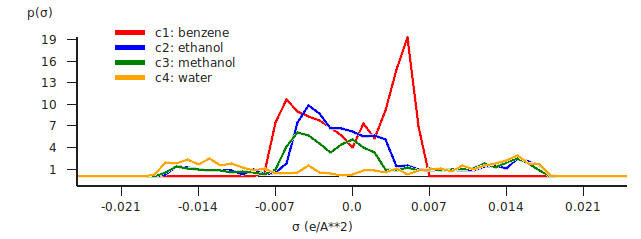
With default settings, if you click in the graph you can zoom (right mouse, or Command-Left (drag up or down)), or translate (left mouse) the graph. If you click in the graph window at the left or below the axes, a popup window will appear in which you can set details for the graph window.
Note that the \(\sigma\)-profile depends on the method (COSMO-RS or COSMO-SAC) that was used in the calculation. Here we have used COSMO-RS. In this case the \(\sigma\)-profile depends on the actual value for r_av (rav ), which is one of the COSMO-RS parameters, see one of the previous steps.
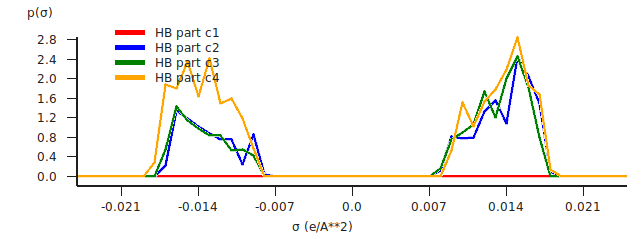
You can also look at the hydrogen bonding part of the \(\sigma\)-profile.
Step 7: Analysis: The sigma potential¶
The sigma potentials (\(\sigma\)-potential) of the four pure compounds will be shown in a graph and in a table in the right part of the window. The \(\sigma\)-potential depends on the temperature of a compound. Here the temperature is set to 25 °C (298.15 K).
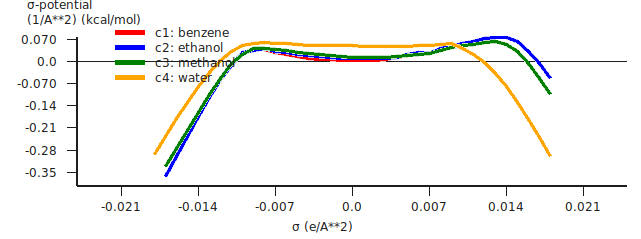
If you double-click on the x-axis in the graph, you can customize the look of the graph on the “Curves” panel in the window that pops up.
Note that the \(\sigma\)-potential is not calculated for values of the COSMO charge density that are non-existent on the COSMO surface of a certain compound.

References