Advanced applications discussed by SCM experts (2020)
In June 2020 we kicked off a series of demonstrations by the SCM developers. We will show tips & trick as well as advanced topics and features in the Amsterdam Modeling Suite. During their short demo / presentation our experts will answer all your questions. Afterwards we’ll also answer your questions in the comments section.
2×2.5h hands-on introduction to the Amsterdam Modeling Suite (virtual winter school on computational chemistry). Slides and inputs available from links in news item.
Materials Modeling with the Amsterdam Modeling Suite
Material properties are ultimately determined at the atomistic level. The Amsterdam Modeling Suite provides a user-friendly, powerful set of computational chemistry tools to study electronic and optical properties, chemical reactivity and mechanical behavior. We introduce our DFT, DFTB and ReaxFF modules and demo the excellent graphical interface. Recent examples from various material science applications in the fields of batteries, organic electronics, and nanotechnology will show how your research can be advanced. The slides and video of the materials modeling web presentation are available.
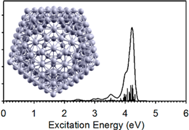
Mauro discusses ADF’s attractive features for calculating optical properties of large nanoparticles. Recent efforts to further increase TDDFT efficiency pave the way for studying even larger nanoparticles, such as large ligand-capped metal clusters. Mauro also discusses how ADF’s spin-orbit-TDDFT and Slater-TS methods can be used to accurately and efficiently calculate X-ray absorption spectra (NEXAFS, XANES). A few hands-on examples are shown to help you with calculating your own UV/VIS and X-ray spectra with ADF.
Literature:
G. Fronzoni, R. De Francesco, and M. Stener, L2,3 edge photoabsorption spectra of bulk V2O5: a two components relativistic time dependent density functional theory description with finite cluster modelJ. Chem. Phys., 137 2240308 (2012) G. Barcaro, L. Sementa, A. Fortunelli, and M. Stener, Optical Properties of Silver Nanoshells from Time-Dependent Density Functional Theory Calculations, J. Phys. Chem. C, 118, 12450-12458 (2014) G. Fronzoni, G. Balducci, R. De Francesco, M. Romeo, and M. Stener, Density Functional Theory Simulation of NEXAFS Spectra of Molecules Adsorbed on Surfaces: C2H4 on Si(100) Case StudyJ. Phys. Chem. C, 116 18910-18919 (2012).
Michele actively develops subsystem DFT for accurately describing charge and excitation energy transfer dynamics in complex systems. Charges, excitons, and spins are localized on particular subsystems within the linear-scaling FDE formulation in ADF. Coupling between these diabatic states are related to charge mobility, energy transfer, charge separation and other processes important in organic electronics and photovoltaics. Michele introduces FDE theory and shows some hands-on examples.
Literature:
P. Ramos and M. Pavanello, Quantifying Environmental Effects on the Decay of Hole Transfer Couplings in BiosystemsJ. Chem. Theory Comput., 10, 2546-2556 (2014). A. Solovyeva, M. Pavanello, and J. Neugebauer Describing long-range charge-separation processes with subsystem density-functional theoryJ. Chem. Phys., 140, 164103 (2014). M. Pavanello, T. van Voorhis, L. Visscher, and J. Neugebauer, An accurate and linear-scaling method for calculating charge-transfer excitation energies and diabatic couplings, J. Chem. Phys. 138, 054101 (2013).
QUILD (QUantum-regions Interconnected by Local Descriptions) features enhanced optimization routines for running multi-level calculations. Marcel introduces the concepts behind QUILD and shows how easy it is to set up subtractive multi-layer QM/MM and QM/QM calcalutions, including how to use QUILD to remove spin contamination from energies, forces and frequencies.
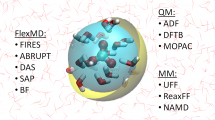
FlexMD is a flexible tool for running multi-scale (QM/MM and QM/QM) molecular dynamics. The python library is interfaced with all modules from the Amsterdam Modeling Suite as well with the Atomistic Simulation Environment and PLUMED. Rosa briefly discussed the different adaptive multi-scale MD schemes implemented in FlexMD and gave live demonstrations on how to set up, run and analyze calculations.
ReaxFF is a powerful tool to study the chemical evolution of large, complex systems. Parametrization becomes increasingly complex for systems with many elements, owing to the many bond-length dependent and cross terms. Eldhose Iype demonstrated the Metropolis Monte Carlo with Simulated annealing (MMC-SA) global optimization technique for fast and automated optimization of ReaxFF parameters given a set of benchmark (DFT) data. He gave a hands-on example and discussed pitfalls, e.g. the need for a good training set that adequately represents the chemical space during the reactive dynamics. New tools that will help parametrization (e.g. converting xyz files to geo format) will soon become available in the development versions of ADF.
Literature:
E. Iype, M. Huetter, A.P.J. Jansen, S.V. Nedea, C.C.M. Rindt, Parameterization of a Reactive Force Field Using a Monte Carlo Algorithm, J. Comp. Chem. 34, 1143-1154 (2013)
An overview of new features and strong points in the 2014 release of the ADF modeling suite, specifically timed for the Australian community. Fedor will cover unique features and functionality of molecular DFT with ADF, periodic DFT with BAND, DFTB, an ReaxFF. There is time for a GUI demonstration, Q&A, and participants watching the webinar at ANU can participate in a hands-on demo session led by NCI’s computational chemistry and ADF expert Dr. Rika Kobayashi. A two-months demo license for the whole ADF modeling suite 2014 will be available at the NCI machines.
Energy decomposition analysis (extended transition state) combined with natural orbitals for chemical valence (EDA-NOCV, ETS-NOCV) is a powerful tool to scrutinize chemical bonding interactions. Mariusz will discuss the underlying philosophy, highlight examples and show how to set up and visualize ETS-NOCV with ADFGUI. Also check out our ETS-NOCV pages for some recent literature examples.
The hybrid method DIM/QM couples the TDDFT excited state calculations of an adsorbed molecule with the atomistic response (Discrete Reaction Field) of a metallic nanoparticle. Lasse presents the methodology, how to set DIM/QM up in ADF, and some recent results for simulating surface-enhanced Raman spectroscopy (SERS). An Accounts of Chemical Research paper was recently published on this topic.