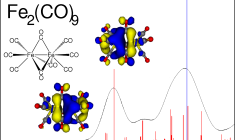
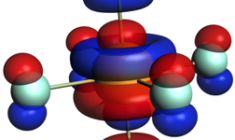
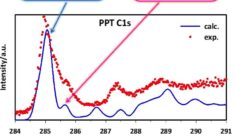
Calculate X-ray absorption spectra: XANES, NEXAFS, XPS
XAS is increasingly used for in situ probing of structures, including catalysts and batteries in operando. X-ray absorption near-edge spectra (XANES) or near-edge X-ray adsorption fine structure spectra (NEXAFS) can be modeled for all elements with ADF. For light elements the DFT-TS approach may give accurate results at little computational effort. Accurate X-ray absorption spectra for heavier elements can be calculated with spin-orbit TDDFT in ADF by restricting the excitations to a selected window of occupied and/or virtual orbitals. X-ray photoelectron spectroscopy (XPS) and Core Electron Binding Energies are easily accessible via core-hole states.
ADF features for calculating X-ray absorption properties
- state-selective excitations, core excitations
- Slater’s Transition State (DFT-TS) and transition potential (TP) for fast XANES
- scalar and spin-orbit relativistic effects
- all-electron Slater basis sets, diffuse functions
- create core-hole states in molecules and periodic systems (BAND)
- model potentials with correct long-range behavior (LB94)
- Ligand Field DFT to include multiplet effects
- X-ray photoelectron spectra (XPS) and CEBE via ΔKohn-Sham
Videos: calculating XPS, XAS with DFT
ADF developer Mauro Stener also gave a web presentation on calculating UV/VIS and X-ray absorption.
Calculating X-ray emission spectra (XES)
The frozen orbital, one-electron ΔDFT approach to calculate X-ray emission energies has been shown to work well for V2C-XES of transition metal complexes. Contributions from quadrupole oscillator strengths can also be included.
User manual and input examples
The powerful ADF features for calculating X-ray spectroscopic properties are export options, meaning that they are not easy to set up via the graphical interface.
The manual entries form a good starting point in combination with the examples shipped with the binaries. Do let us know at support@scm.com if you have any problems or questions.
- X-ray absorption: manual, video: state selection, modified excitation range, orbital energy differences (TS, TP)
- Tutorial: NEXAFS calculation
- Tutorial: XPS or core-electron binding energies (see also: ppt and input files, video)
- X-ray emission: manual
- ADF examples
- BAND example: electron hole in Si
- Webinar by Harry Ramanantoanina on Ligand Field DFT (LFDFT) a practical tool for coordination chemistry (slides, video, LFDFT inputs and step-by-step instructions) includes XAS examples