HCN Isomerization Reaction¶
This tutorial consists of several steps to study the isomerization reaction of HCN:
- Geometry optimization
- Linear transit
- Quick frequency calculation
- Transition state search
- Accurate frequency calculation
- IRC
Step 1: Prepare the HCN molecule¶
- Start ADFjobsClick on the Tutorial folder iconStart ADFinput using the SCM → New Input menu commandDraw an HCN molecule (first the N, next the C and finally an H atom)Select the C-N bond and make it a triple-bondPre-optimize the geometry
You should get a linear molecule:

- Select the Geometry Optimization taskSelect the DZP basis setSelect File → Run, give it the name HCN_GO
The geometry will be optimized, using a DZP basis set.
- Click “Yes” when asked to read new coordinates from the HCN_GO.t21 fileCheck the C-N and C-H distances
They should be about 115 and 108 pm (1.15 and 1.08 Angstrom), respectively.
- Write down the value of the bonding energy printedat the end of the calculation in the ADFtail window
Step 2: Create a rough approximation for the transition state geometry¶
The HCN molecule has an CNH isomer. There is an energy barrier between these two states. We are going to find the transition state and calculate its height.
To find a better starting point for the transition state search we will perform a linear transit calculation as a rough approximation of the reaction path. We will vary the H-C-N angle in steps between 40 and 140 degrees and let ADF optimize bond lengths at each angle.
To set up the linear transit calculation:
- Select the Linear Transit taskClick on the
 button next to task to go to the ‘Geometry Constraints and Scan’ panelSelect all the atoms
button next to task to go to the ‘Geometry Constraints and Scan’ panelSelect all the atoms
You should see ‘+ N(1) C(2) H(3) (angle)’ note in the right panel now:

- Click the ‘+’ button to add the angle constraint
Now ‘- N(1) C(2) H(3)’ and the two fields as limits for the degree parameter appear.
- Enter ‘140’ and ‘40’ in the ‘degrees’ fields
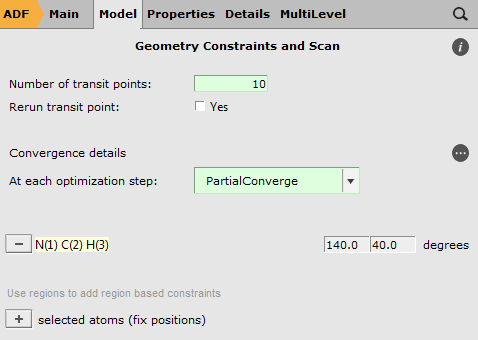
ADF will have trouble running the current set up because the HCN molecule is perfectly linear. So we will help ADF by changing the angle to 140 degrees, the same as the first point of the LT scan.
- Use the slider to change the HCN angle to 140 degrees

The set up is complete. Now we will run the LT calculation, but we will save it with a new name as we wish to keep the results of the HCN_GO calculation:
- Use File → Save As to save the file as ‘HCN_LT’Run the calculation
Running might take a few minutes. When the run is finished:
- Click “Yes” when asked to read new coordinates
You will see the last geometry, close to CNH. To see how geometry was changing during the LT run, use ADFmovie:
- Select the SCM → Movie commandSelect all atoms (use shift-drag to make a rectangle around the atoms)Select the Graph → Energy commandSelect the Graph → Distance, Angle, Dihedral command (to get the angle graph, as three atoms are still selected)Use the View → Converged Geometries Only commandZoom in to get a better view of the moleculePress the Play button (the right-pointing triangle in the controls at the left bottom of the ADFmovie window)
You will see the hydrogen atom moving from C to N. You will also see a graph of the energy as function of the LT steps. As the movie is playing a dot shows the corresponding position in the graph.
Somewhere along the path, there is a transition state we are looking for. Remember that you needed to use the ‘Optimized Geometries Only’ command to filter out all the intermediate geometry step, so that you get only the converged geometries for each LT step.
- In the graph, click (without moving!) on the top of the energy graphAlternatively, use the arrow keys (cursor keys) to move between different steps, or use the sliderCheck which geometry has the maximum energy
You should find that at around an angle of 60-75 degrees the maximum energy is reached. This is Frame 6 (the 7th LT step):

You can find this information also in the output file:
- Select the SCM → Output menu commandIn the ADFoutput window select the Other Properties → ‘LT Path command
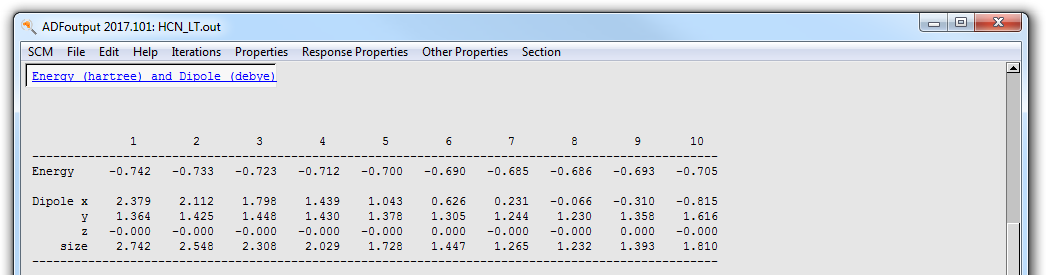
You will see that indeed the geometry number 7 (corresponding to Frame 6 in ADFmovie) has the highest energy. In this particular example the choice of the angle is not very important, but in general you will always want to get the best approximation for the transition state available.
We will now prepare the search for the transition state starting from this geometry:
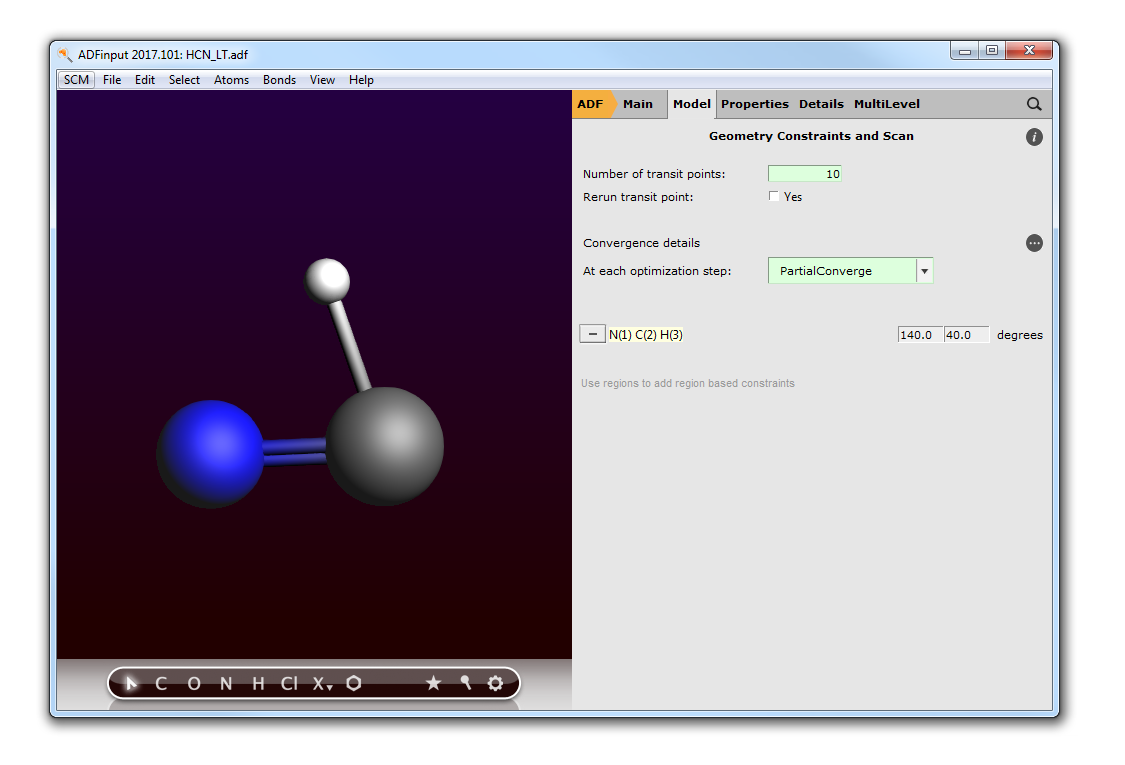
- Click in the ADFmovie windowMake sure frame 6 is selectedUse the File → Update Geometry In Input command
The geometry of HCN in your ADFinput window will be updated to match the geometry currently selected in the ADFmovie window:
Step 3: Finding the transition state: prepare approximate Hessian¶
In general, it is important to have a good starting Hessian with one imaginary frequency when performing a TS search. We are going to create such a Hessian by doing a quick frequencies calculation:
- Select the Frequencies task (from the Main panel)Make sure the Numerical quality is set to NormalSave the molecule as ‘HCN_Freq1’ (Save As)Run the calculation
The frequency calculation is now in progress and will run very fast. When it has finished:
- Select the SCM → Spectra command

If everything was done correctly, you should see a spectrum with three peaks: two in the range of 2000 - 3000 1/cm and one peak in the range of negative or very low positive values. A negative frequency value actually means that it is an imaginary frequency.
- Click with your mouse on the peak corresponding to the imaginary frequency
The normal mode corresponding to that frequency will show on the left side. Check that the frequency indeed corresponds to the H atom moving parallel to the C-N bond.
Step 4: Search for the transition state¶
The result file, HCN_Freq1.t21, has an initial geometry for our transition state search. It also contains a Hessian matrix (produced with the frequencies calculation) that can be used to kick-start the TS procedure.
- Bring ADFinput with the HCN_Freq1 calculation to the foregroundSelect the Transition State taskctrl/cmd-F and search for ‘restart’, select the ‘Files (Restart)’ panelClick the file select button (looks like a folder) in front of the empty ‘Restart file:’ fieldSelect the HCN_Freq1.t21 file and click ‘Open’:

- Save the set up as HCN_TS (Save As)Run the calculation
After the calculation has finished (again very fast), you will be asked to read the new geometry from the results file HCN_TS.t21:
- Answer “Yes” to import the latest geometryMake a note of the bond energy for the transition state (visible in the logfile)
ADFinput will now display the transition state geometry.
If you compare the bond energy with the bond energy of the optimized HCN molecule from the first calculation, the difference should be about 1.9 eV. Also check that the geometry makes sense: the C-H and C-N distances should be around 1.20 and 1.19 Angstrom and the H-C-N angle should be about 70 degrees.
Step 5: Calculating frequencies at the transition state¶
After every transition state search it is good practice to verify that you indeed have one and only one imaginary frequency. For this we will repeat the frequency calculation at the TS geometry:
- Make sure you have HCN_TS open in ADFinputSelect the Frequencies task (from the ‘Main’ panel)Save with name HCN_Freq2Run
The calculation is running, should not take much time. After the calculation has finished:
- Select the SCM → Spectra command
You will be presented with an IR spectrum of the molecule featuring three bands roughly located at 2550, 2050, and (imaginary) -1040 1/cm.
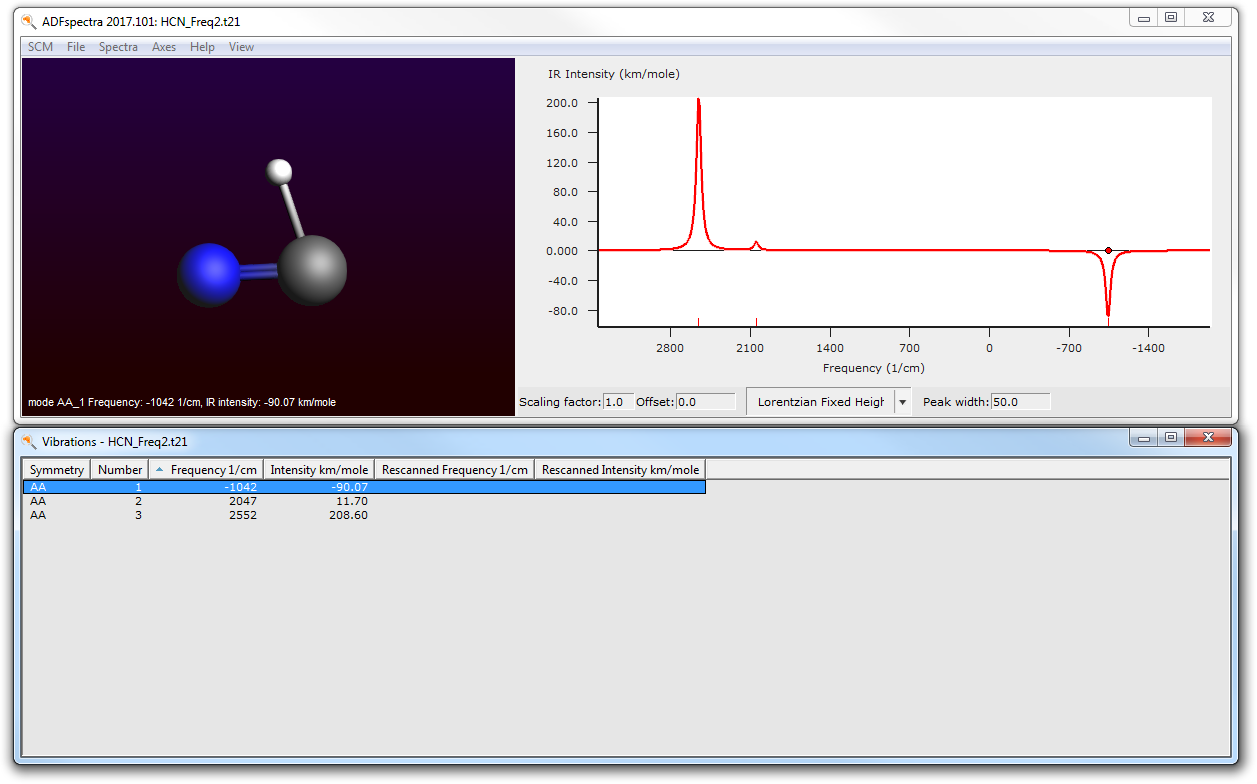
- Click on the band at -1040
The visualization of the normal mode corresponding to this frequency should make it clear that this is indeed the reaction coordinate we are studying.
Step 6: Following the reaction coordinate¶
ADF can follow the minimum-energy path from the transition state to one or the other product. The method used in ADF for this is called Intrinsic Reaction Coordinate (IRC). You may want to skip this part as the calculation might take some time to complete.
- Bring HCN_Freq2 in ADFinput to the frontSelect the IRC taskGo to the ‘Intrinsic Reaction Coordinate (IRC)’ panel (in Model, or via the search, or by clicking on the button)
This panel allows you to specify various parameters for the IRC method. The most important parameter is the direction to follow. The choice is more or less arbitrary. By choosing “Forward path” or “Backward path” will lead you to one or the other product but it’s hard to tell which of the two in what case. We will calculate both paths at once, so we do not need to change the default.
Another option is the ‘Rerun IRC points’. When checked, after the IRC calculation has finished an extra single point calculation will be performed for each converged points, and the results will be saved. This will allow you to observe in more detail what happens along the IRC path, for example how orbitals change. In this tutorial, lets show how this work, so turn that feature on:
- Check the ‘Rerun IRC points’ checkbox
We also want to make sure all optimizations converge, so lets increase the maximum number of iterations to 50:
- Click on the after Convergence detailsIn the Number of geometry iterations field specify 50Save as HCN_IRCRun
After some minutes the calculation will finish. You can use ADFmovie to view the IRC path. Of course you need again to make sure to show the converged geometries only:
- Select the SCM → Movie commandSelect the View → Converged Geometries Only commandClick OK when the warning message appears (about the changed order of steps)Select the Graph → Energy commandSelect all atomsSelect the Graph → Distance, Angle, Dihedral command
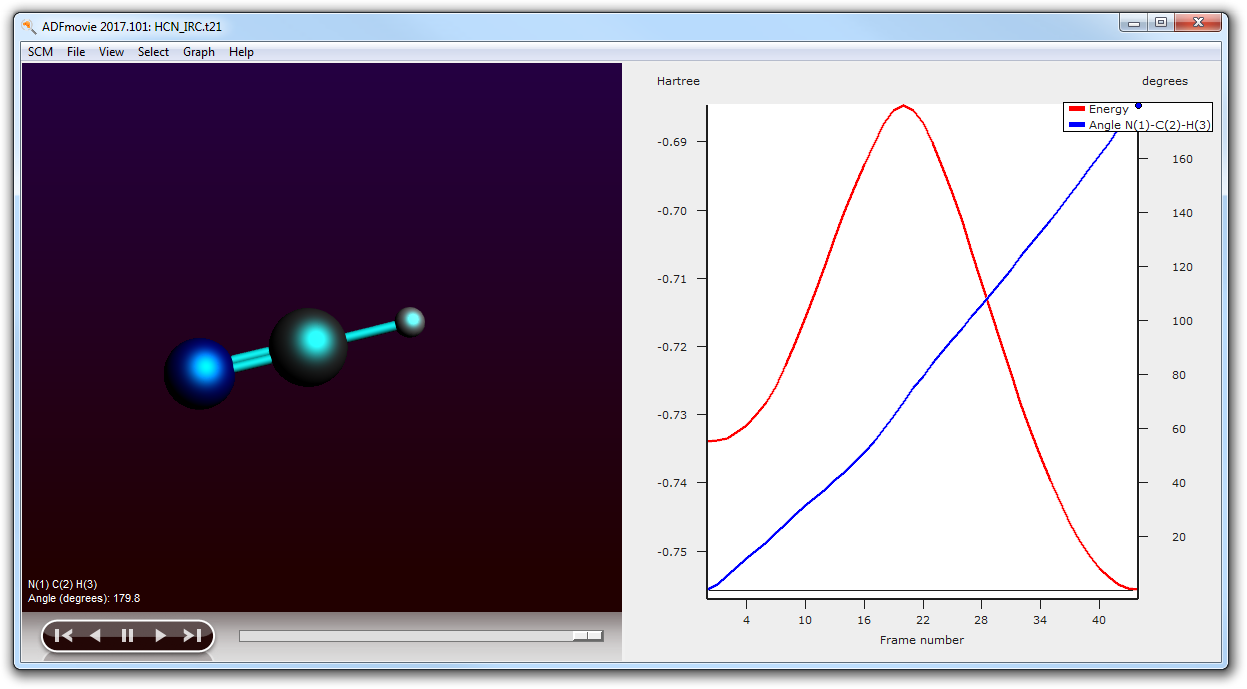
From this movie you can see the IRC path, and the energies at the most interesting points. As we have calculated the forward and backward path in one run, the order of the forward path needs to be reversed to get a proper energy graph.
You can also examine some properties along the IRC path by studying the output file:
- Select SCM → Output (this might take a while, be patient)Use the Other Properties → IRC Path menu command
You will see a table with the properties along the forward path. To get the backwards path:
- Click on the blue header ‘Dist from TS ...’
The output browser should jump to the next section with that header, which is the table for the backward IRC path.
Step 7: Following orbitals along the IRC: reporting from .t21 files¶
In the previous IRC calculation you had activated the Rerun IRC points option. As a result the data for all the converged IRC points is also available.
- Bring ADFjobs to the foregroundClick on the triangle for the HCN_IRC job to see the job files

If you wish you can select the .t21 file of interest, and use the GUI tools (ADFview, ADFlevels, KFBrowser) to examine the results for that particular IRC point in detail. You can also use the Report tool to generate an overview:
- Job → Open .results to open the .results in ADFjobsSelect all sp_... jobs (click on the first, shift click on the last)Tools → Build Standard ReportSave the job with a name like ‘HCN IRC results’Wait until the report generation is ready,the ADFjobs status line at the top will give an indication of the progress
Now you should have a nice overview of the detailed results, as set up in the report options, for all of the IRC points:

Another example of using the Report tool can be found in NH3 Basis Set tutorial.
Step 8: Following orbitals for the LT afterwards: generating jobs for many geometries¶
For Linear Transit runs you also might want to follow orbitals, or check other properties for the converged points. You can do this in exactly the same way as for the IRC run (thus, by activating the Rerun LT points option). However, this option was not activated.
There is an easy way to generate the information for the converged points afterwards as well. Note that this would also work after an IRC calculation instead of a LT calculation.
- Activate ADFjobsSelect the HCN_LT jobTools → PrepareIn the Prepare window that appears:In the “Use coordinates from” section, press the “+” and select “ADF result file()s (.t21)”Select the HCN_LT.t21 results file, and click OpenIn the “Use the input options section”, press the “+” and select “Run Type → SinglePoint”

- Click OKSelect all the SP.HCN jobs that have been generatedJob → RunWait for the calculations to finish
Now we can use the Report Tool to generate an overview, just as we just did for the IRC calculations:
- Select all SP.HCN_LT...jobs (click on the first, shift click on the last)Tools → Build Standard ReportSave the job with a name like ‘HCN LT results’Wait until the report generation is ready,the ADFjobs status line at the top will give an indication of the progress
Now you should have a nice overview of the detailed results, as set up in the report options, for all of the LT points:

Finally, at the end of this tutorial you will have many open windows. To close all ADF-GUI related windows at once, you may use the SCM → Quit All command.