PEDA for Spin Unrestricted Calculations¶
This tutorial will teach you how to:
- perform a periodic energy decomposition analysis (PEDA) for extended systems with the BAND-GUI, where the bond is described by open-shell, spin unrestricted fragments
- where to look for the results
Step 1: Start ADFinput¶
Start ADFinput in a clean directory. (according to Step 1 of the Getting Started with BAND)
Step 2: Set up the system - Ethane¶
An easy and non-timeconsuming testsystem is the C-C bond analysis in an ethane molecule. Here, two methyl radicals with opposite spin-polarization do interact to form the shared electron C-C bond. Hence, the fragments and the PEDA calculation have to be carried out as unrestricted DFT calculations.
You can copy-paste the following geometry information into the GUI directly.
C 1.54081631 0.00000000 0.00000000
H 1.90061013 0.71448558 0.72714386
H 1.90061011 -0.98692880 0.25534173
H 1.90084793 0.27238885 -0.98228957
C 0.00000000 0.00000000 0.00000000
H -0.35953041 -0.27213306 0.98136715
H -0.36080772 0.98640196 -0.25411759
H -0.36080775 -0.71466441 -0.72582318
VEC1 10.00000000 0.00000000 0.00000000

Step 3: Running the PEDA calculation¶
Step 3a: Setting up the fragments¶
To run the PEDA you have to define the fragments. Therefore switch to Regions menu.
- Panel bar Model → Regions
- Select one methyl fragment and add a new region by clicking on the ‘+’ button (or Ctrl+G).
- Then select the other methyl fragment and add a new region by clicking on the ‘+’ button (or Ctrl+G).
- You may want to rename “Region_1” to “H3C_A” and “Region_2” to “H3C_B”. (optional)

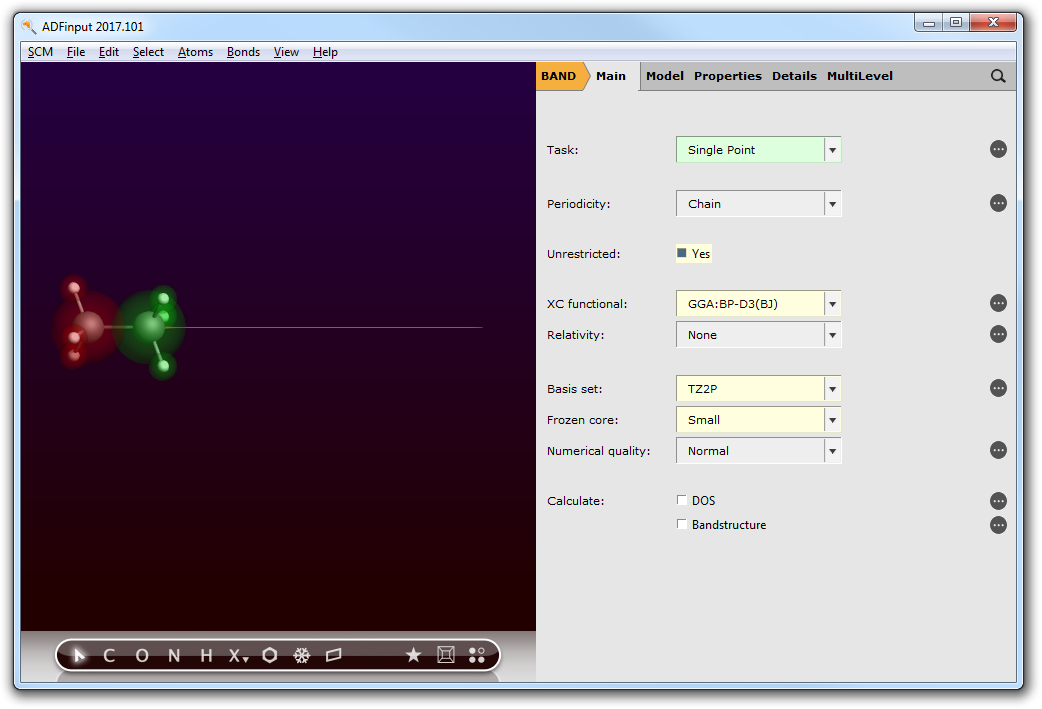
Step 3b: Details for the calculation¶
Go to the Main menu,
- Panel bar Main
and change the calculation setup (XC functional, basis set, frozen core, numerical quality and unrestricted calculation) according to the following picture.
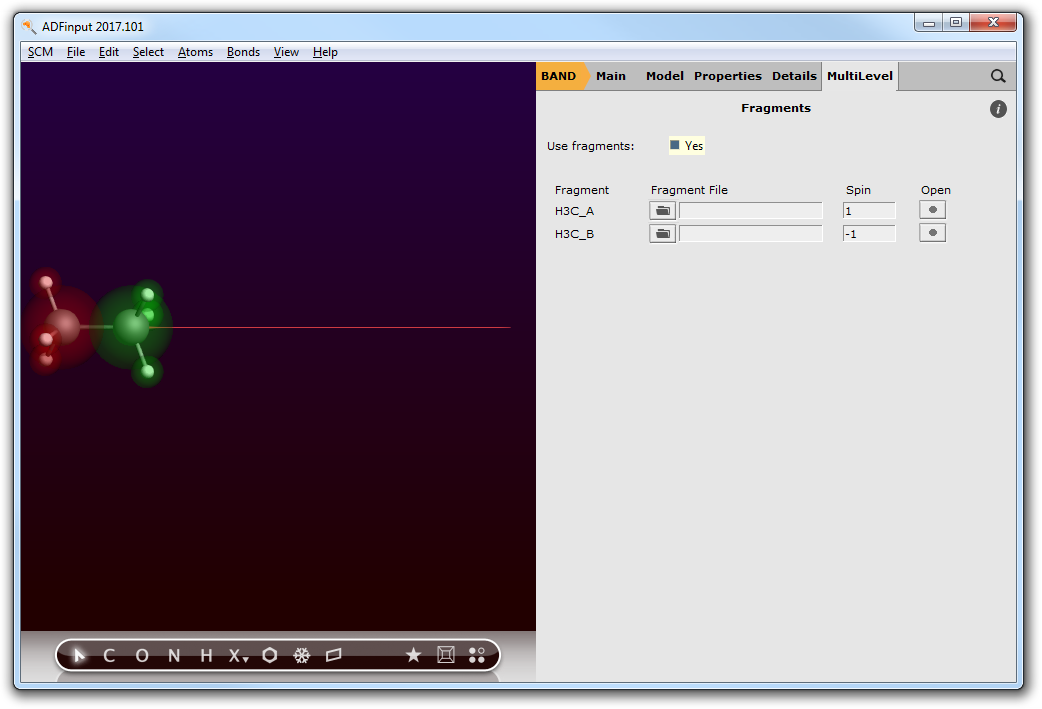
Step 3c: Enabling the PEDA¶
Go to the Fragments menu,
- Panel bar MultiLevel → Fragments
- Check the “Use fragments” box. This will trigger the PEDA.
- Define the spin polarization of the fragments. One shall be +1 (excess of 1 electron with spin up) and the other -1 (excess of 1 electron with spin down).
Step 3d: Save and run the calculation¶
Now you can save and run the calculation.
- File → Save, give it a name and press Save.File → Run
Step 4: Checking the results¶
After the calculations of the fragments and the PEDA finished you can look for the PEDA results. Therefore, open the “Output” using the SCM dropdown menu.
- SCM → Output
You can jump to the ‘PEDA Energy Terms’ via the corresponding button in the ‘Properties’ dropdown menu.
- Properties → PEDA Energy Terms
Reference results:

In addition to these energy terms the summed preparation energies of the fragments and the (negative) bond dissociation energy are usually given. Therefore you have to calculate the energy difference between the electronically and structurally relaxed fragments (which can be accessed by a geometry optimization of the separated fragments) and the promoted fragments (which are already calculated and used for the PEDA). Adding this energy differences, which are equal to the preparation energy, to the interaction energy will give you the negative bond dissociation energy.