Active Learning: Conformer Training with CREST¶
Build a custom M3GNet model for conformer ranking by combining Simple Active Learning, CREST metadynamics, and additional reference conformers in Python.
Trained model: M3GNet, starting from the Universal Potential (UP)
Reference method: GFN-1xTB (DFTB engine)
System: Organic molecule with SMILES code OC(CC1c2ccccc2Sc2ccccc21)CN1CCCC1
Problem: M3GNet-UP-2022 is not reliable for conformer stability prediction:
Solution: Retraining the model gives better agreement.
Expected duration: This notebook takes about 24 hours to run on the CPU. This includes both the active learning and the production simulations to generate new conformers.
When running active learning it’s usually a good idea to start off “simple” and make the system/structures gradually more complicated.
For getting a model which predicts conformers accurately, we may take the following approach:
first train a potential at slightly above room temperature with NVT MD
continue training a second potential using CREST metadynamics
generate conformers with the previous model and also train to those
Initial imports¶
import scm.plams as plams
import scm.params as params
from scm.simple_active_learning import SimpleActiveLearningJob
import matplotlib.pyplot as plt
import os
import numpy as np
from typing import List
from scm.conformers import ConformersJob
from scm.conformers.plams.plot import plot_conformers
plams.init()
PLAMS working folder: /path/to/plams_workdir
Create the initial structure¶
molecule = plams.from_smiles("OC(CC1c2ccccc2Sc2ccccc21)CN1CCCC1", forcefield="uff")
molecule.delete_all_bonds()
for at in molecule:
at.properties = plams.Settings()
plams.plot_molecule(molecule)
starting_structure = molecule
Reference engine settings¶
fast_ref_s = plams.Settings()
fast_ref_s.input.DFTB.Model = "GFN1-xTB"
slow_ref_s = plams.Settings()
slow_ref_s.input.ADF.Basis.Type = "TZP"
slow_ref_s.input.ADF.Basis.Core = "None"
slow_ref_s.input.ADF.XC.Hybrid = "B3LYP"
slow_ref_s.input.ADF.XC.DISPERSION = "GRIMME3 BJDAMP"
Change to slow_ref_s to train to B3LYP-D3(BJ) instead:
ref_s = fast_ref_s.copy()
# ref_s = slow_ref_s.copy()
perform_expensive_tests = ref_s == fast_ref_s
Problem statement: Generate a few conformers with M3GNet-UP-2022 and Score with reference method¶
m3gnet_up_s = plams.Settings()
m3gnet_up_s.input.MLPotential.Model = "M3GNet-UP-2022"
generate_conformers_m3gnet_up_job = ConformersJob(
name="generate_conformers_m3gnet_up", molecule=starting_structure
)
generate_conformers_m3gnet_up_job.settings.input.ams.Task = "Generate"
generate_conformers_m3gnet_up_job.settings.input.ams.Generator.Method = "RDKit"
generate_conformers_m3gnet_up_job.settings.input.ams.Generator.RDKit.InitialNConformers = 40
# generate_conformers_m3gnet_up_job.settings += crest_al_job.results.get_production_engine_settings()
generate_conformers_m3gnet_up_job.settings += m3gnet_up_s
# generate_conformers_m3gnet_up_job.settings.runscript.nproc = 1 # run in serial, useful if you run out of memory when running M3GNet on the GPU
generate_conformers_m3gnet_up_job.run();
[13.03|09:15:30] JOB generate_conformers_m3gnet_up STARTED
[13.03|09:15:30] JOB generate_conformers_m3gnet_up RUNNING
[13.03|09:17:10] JOB generate_conformers_m3gnet_up FINISHED
[13.03|09:17:10] JOB generate_conformers_m3gnet_up SUCCESSFUL
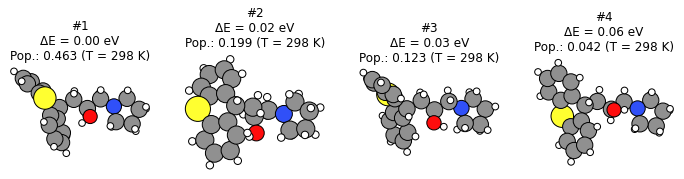
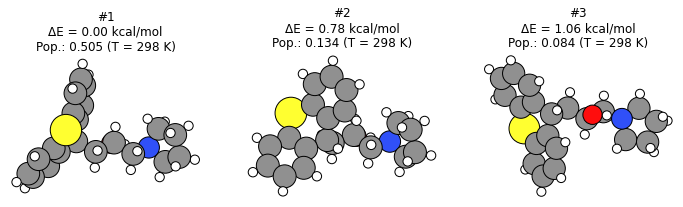
Show the 4 most stable conformers with M3GNet-UP-2022:
plot_conformers(generate_conformers_m3gnet_up_job, 4, unit="eV");
Score the generated conformers with the reference method:
score_conformers_ref_job = ConformersJob(name="score_conformers_ref")
score_conformers_ref_job.settings.input.ams.Task = "Score"
score_conformers_ref_job.settings.input.ams.InputConformersSet = (
generate_conformers_m3gnet_up_job.results.rkfpath()
)
score_conformers_ref_job.settings.input += ref_s.input
score_conformers_ref_job.run();
[13.03|09:17:10] JOB score_conformers_ref STARTED
[13.03|09:17:10] JOB score_conformers_ref RUNNING
[13.03|09:17:16] JOB score_conformers_ref FINISHED
[13.03|09:17:16] JOB score_conformers_ref SUCCESSFUL
plot_conformers(score_conformers_ref_job, 4, unit="eV");
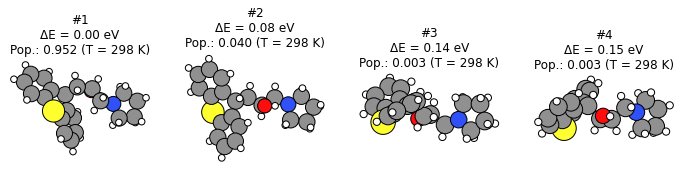
Here we can see that the ordering of conformers at the reference level is completely different compared to M3GNet-UP-2022.
The Score job reorders the conformers. To compare the energies more precisely, we can use the minimum pairwise RMSD (which should be 0) to identify how the order of conformers change:
def get_pairwise_rmsds(
molecules_ref: List[plams.Molecule], molecules_pred: List[plams.Molecule]
) -> np.ndarray:
"""Returns a len(molecules_ref)*len(molecules_pred) numpy array with pairwise rmsds (in angstrom) between the structures"""
rmsds = np.zeros((len(molecules_ref), len(molecules_pred)))
for i, ref_mol in enumerate(molecules_ref):
for j, pred_mol in enumerate(molecules_pred):
rmsds[i, j] = plams.Molecule.rmsd(ref_mol, pred_mol)
return rmsds
def get_reordering(rmsds: np.ndarray) -> np.ndarray:
"""
rmsds: numpy array with shape len(molecules_ref)*len(molecules_pred)
Returns a len(molecules_ref) integer numpy array.
The first element is the index (0-based) in molecules_pred corresponding to the first reference molecule, etc.
"""
rmsds = get_pairwise_rmsds(molecules_ref, molecules_pred)
reordering = np.argmin(rmsds, axis=1)
return reordering
def print_reordering_table(
molecules_ref, molecules_pred, energies_ref, energies_pred, ax=None
):
"""This functions prints the reordering table including relative energies. It also plots the predicted relative energies versus the reference relative energies"""
print(f"Ref_i Pred_i RMSD Ref_dE Pred_dE")
x, y = [], []
rmsds = get_pairwise_rmsds(molecules_ref, molecules_pred)
reordering = get_reordering(rmsds)
rmsd_threshold = 0.7 # angstrom, for printing/plotting points
for ref_i, pred_i in enumerate(reordering):
rmsd = rmsds[ref_i, pred_i]
ref_relative_e = energies_ref[ref_i]
pred_relative_e = energies_pred[pred_i] - energies_pred[reordering[0]]
if rmsd <= rmsd_threshold:
print(
f"{ref_i:6d} {pred_i:6d} {rmsd:4.1f} {ref_relative_e:7.2f} {pred_relative_e:7.2f}"
)
x.append(ref_relative_e)
y.append(pred_relative_e)
else:
print(f"{ref_i:6d} {pred_i:6d} rmsd > {rmsd_threshold:.1f} ang.")
if ax is None:
_, ax = plt.subplots()
m, M = np.min([np.min(x), np.min(y)]), np.max([np.max(x), np.max(y)])
ax.plot(x, y, ".")
ax.plot([m, M], [m, M], "-")
ax.set_xlabel("ref. deltaE (eV)")
ax.set_ylabel("pred. deltaE (eV)")
return ax
In the below table the energies are given with respect to the structure that had the lowest reference energy. This is thus different compared to the first image above, where the predicted (m3gnet-up-2022) relative energies were given to predicted lowest energy conformer.
molecules_ref = score_conformers_ref_job.results.get_conformers()
energies_ref = score_conformers_ref_job.results.get_relative_energies(unit="eV")
molecules_pred = generate_conformers_m3gnet_up_job.results.get_conformers()
energies_pred = generate_conformers_m3gnet_up_job.results.get_relative_energies(
unit="ev"
)
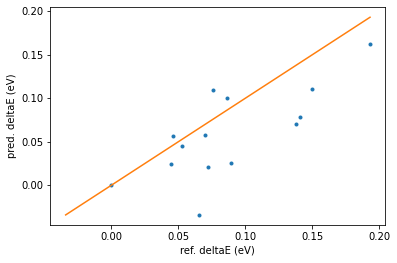
ax = print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred)
ax.set_title("M3GNet-UP-2022 relative to reference method");
Ref_i Pred_i RMSD Ref_dE Pred_dE
0 5 0.0 0.00 0.00
1 3 0.0 0.08 -0.01
2 19 0.0 0.14 0.07
3 20 0.0 0.15 0.08
... output trimmed ....
20 12 0.0 0.56 0.04
21 24 0.0 0.60 0.17
22 8 0.0 0.68 0.02
23 13 0.0 0.75 0.04
24 1 0.0 0.97 -0.05
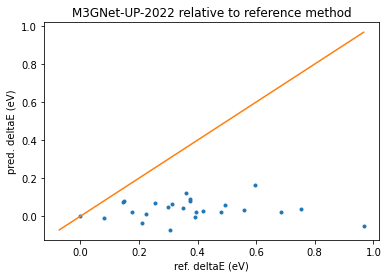
Here we see that there is no correlation between the relative stabilities calculated with M3GNet-UP-2022 and the reference method.
Example: The most stable conformer with the reference method (Ref_i=0) corresponds to the tenth most stable conformer with M3GNet-UP-2022 (Pred_i=10)
Example 2: The second most stable conformer with the reference method (Ref_i=1) should be 0.08 eV less stable than the ref_i=0 conformer, but with M3GNet-UP-2022 method it is actually 0.01 eV more stable!
Optimize a few conformers for initial reference data¶
Conformers are local minima on the potential energy surface. The Active Learning MD will sample off-equilibrium structures. So let’s make sure that there are at least some local minima in the training set by optimizing some of the generated conformers.
Here, we loop over the conformers and run GeometryOptimization jobs. This produces output files in the normal AMS format that can easily be imported into ParAMS
max_N = min(6, len(molecules_pred)) # at most 6 optimizations
opt_s = plams.Settings()
opt_s.input.ams.Task = "GeometryOptimization"
opt_s.input.ams.GeometryOptimization.Convergence.Quality = "Basic"
opt_jobs = [
plams.AMSJob(settings=opt_s + ref_s, name=f"opt_{i}", molecule=mol)
for i, mol in enumerate(molecules_pred[:max_N])
]
for opt_job in opt_jobs:
opt_job.run()
[13.03|09:17:18] JOB opt_0 STARTED
[13.03|09:17:18] JOB opt_0 RUNNING
[13.03|09:17:28] JOB opt_0 FINISHED
[13.03|09:17:28] JOB opt_0 SUCCESSFUL
[13.03|09:17:28] JOB opt_1 STARTED
... output trimmed ....
[13.03|09:18:08] JOB opt_4 SUCCESSFUL
[13.03|09:18:08] JOB opt_5 STARTED
[13.03|09:18:08] JOB opt_5 RUNNING
[13.03|09:18:14] JOB opt_5 FINISHED
[13.03|09:18:14] JOB opt_5 SUCCESSFUL
Now import the data into a ParAMS results importer and store in the directory my_initial_reference_data:
yaml_dir = os.path.abspath("my_initial_reference_data")
ri = params.ResultsImporter(
settings={"units": {"energy": "eV", "forces": "eV/angstrom"}}
)
for opt_job in opt_jobs:
ri.add_singlejob(opt_job, properties=["energy", "forces"], task="SinglePoint")
ri.store(yaml_dir, backup=False)
['/home/hellstrom/testnb/conformers/my_initial_reference_data/job_collection.yaml',
'/home/hellstrom/testnb/conformers/my_initial_reference_data/results_importer_settings.yaml',
'/home/hellstrom/testnb/conformers/my_initial_reference_data/training_set.yaml']
Simple Active Learning setup¶
Molecular dynamics settings (temperature ramp)¶
nvt_md_s = plams.AMSNVTJob(
nsteps=20000,
timestep=0.5,
temperature=(270, 350, 350),
tau=100,
thermostat="Berendsen",
).settings
ParAMS machine learning settings¶
ml_s = plams.Settings()
ml_s.input.ams.MachineLearning.Backend = "M3GNet"
ml_s.input.ams.MachineLearning.CommitteeSize = 1
ml_s.input.ams.MachineLearning.M3GNet.Model = "UniversalPotential"
ml_s.input.ams.MachineLearning.MaxEpochs = 200
Active learning settings¶
Conformer search of a single molecule is quite simple, so we can expect the ML method to perform quite well. We therefore decrease the success criteria thresholds a bit compared to the default values, to ensure that we get accurate results.
Since we will immediately continue with another active learning loop, we disable the “RerunSimulation” as we are not interested in the MD simulation per se.
al_s = plams.Settings()
al_s.input.ams.ActiveLearning.Steps.Type = "Geometric"
al_s.input.ams.ActiveLearning.Steps.Geometric.Start = 10
al_s.input.ams.ActiveLearning.Steps.Geometric.NumSteps = 8
al_s.input.ams.ActiveLearning.InitialReferenceData.Load.Directory = yaml_dir
al_s.input.ams.ActiveLearning.InitialReferenceData.Generate.M3GNetShortMD.Enabled = (
"Yes"
)
al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.002
al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = 0.30
al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
Initial structure¶
Let’s start with the lowest-energy conformer according to the reference method:
starting_structure_al = molecules_ref[0]
# The bonds are not used, so delete them to make the input file less confusing:
starting_structure_al.delete_all_bonds()
Complete job¶
settings = ref_s + nvt_md_s + ml_s + al_s
job = SimpleActiveLearningJob(
settings=settings, molecule=starting_structure_al, name="sal"
)
print(job.get_input())
ActiveLearning
AtEnd
RerunSimulation False
End
InitialReferenceData
... output trimmed ....
H 3.4863964364 -2.0479923169 -1.0050050526
H 4.0606739540 -1.7926476557 0.6541872002
End
End
Run the simple active learning job¶
job.run(watch=True);
[13.03|09:18:15] JOB sal STARTED
[13.03|09:18:15] JOB sal RUNNING
[13.03|09:18:17] Simple Active Learning 2024.101, Nodes: 1, Procs: 64
[13.03|09:18:21] Composition of main system: C20H23NOS
[13.03|09:18:21] All REFERENCE calculations will be performed with the following DFTB engine:
... output trimmed ....
[13.03|10:33:12] Active learning finished!
[13.03|10:33:12] Goodbye!
[13.03|10:33:13] JOB sal FINISHED
[13.03|10:33:13] JOB sal SUCCESSFUL
Final structure from MD simulation¶
new_structure = job.results.get_main_molecule()
plams.plot_molecule(new_structure, rotation="-5x,5y,0z")
Second active learning job: CREST metadynamics¶
Here we set Steps.Type = “Linear” to run reference calculations every 2000 MD steps.
nsteps = 20000
crest_md_s = plams.Settings()
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.Height = 0.138
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.NGaussiansMax = 50
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.NSteps = 200
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.Width = 0.62
crest_complete_md_s = plams.AMSMDJob(
molecule=new_structure,
nsteps=nsteps,
settings=crest_md_s,
tau=10, # small time constant
thermostat="NHC",
temperature=300,
timestep=0.5,
samplingfreq=20,
).settings
# job = SimpleActiveLearningJob.load_external(plams.config.default_jobmanager.workdir + "/sal.002")
crest_ml_s = ml_s.copy()
crest_ml_s.input.ams.MachineLearning.LoadModel = (
job.results.get_params_results_directory()
)
crest_ml_s.input.ams.MachineLearning.Target.Forces.MAE = 0.04
crest_ml_s.input.ams.MachineLearning.MaxEpochs = 200
crest_al_s = plams.Settings()
crest_al_s.input.ams.ActiveLearning.Steps.Type = "Linear"
crest_al_s.input.ams.ActiveLearning.Steps.Linear.Start = 500
crest_al_s.input.ams.ActiveLearning.Steps.Linear.StepSize = 2000
crest_al_s.input.ams.ActiveLearning.InitialReferenceData.Load.FromPreviousModel = "Yes"
crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.002
crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Total = 0.010
# because we do not set Normalization, the above Energy criteria are energies per atom
# crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Normalization =
crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = (
0.30
)
crest_al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
crest_al_s.input.ams.ActiveLearning.MaxReferenceCalculationsPerAttempt = 3
crest_al_job = SimpleActiveLearningJob(
name="crest_al",
settings=ref_s + crest_complete_md_s + crest_ml_s + crest_al_s,
molecule=new_structure,
)
crest_al_job.run(watch=True);
[13.03|10:33:13] JOB crest_al STARTED
[13.03|10:33:13] JOB crest_al RUNNING
[13.03|10:33:16] Simple Active Learning 2024.101, Nodes: 1, Procs: 64
[13.03|10:33:19] Composition of main system: C20H23NOS
[13.03|10:33:19] All REFERENCE calculations will be performed with the following DFTB engine:
... output trimmed ....
[13.03|14:11:32] Active learning finished!
[13.03|14:11:32] Goodbye!
[13.03|14:11:33] JOB crest_al FINISHED
[13.03|14:11:33] JOB crest_al SUCCESSFUL
# crest_al_job = SimpleActiveLearningJob.load_external("plams_workdir.003/crest_al")
new_retrained_model_settings = (
crest_al_job.results.get_params_job().results.get_production_engine_settings()
)
Generate conformers with the retrained M3GNet model and score with reference method¶
def generate_and_score(
molecule: plams.Molecule,
gen_name: str,
gen_settings: plams.Settings,
score_name: str,
score_settings: plams.Settings,
):
generate_job = ConformersJob(name=gen_name, molecule=molecule)
generate_job.settings.input.ams.Task = "Generate"
generate_job.settings.input.ams.Generator.Method = "RDKit"
generate_job.settings.input.ams.Generator.RDKit.InitialNConformers = 40
generate_job.settings.input += gen_settings.input
generate_job.run()
score_job = ConformersJob(name=score_name)
score_job.settings.input.ams.Task = "Score"
score_job.settings.input.ams.InputConformersSet = generate_job.results.rkfpath()
score_job.settings.input += score_settings.input
score_job.run()
molecules_gen = generate_job.results.get_conformers()
energies_gen = generate_job.results.get_relative_energies(unit="eV")
molecules_score = score_job.results.get_conformers()
energies_score = score_job.results.get_relative_energies(unit="ev")
return (
generate_job,
molecules_gen,
energies_gen,
score_job,
molecules_score,
energies_score,
)
(
generate_conformers_m3gnet_retrained_job,
molecules_pred,
energies_pred,
_,
molecules_ref,
energies_ref,
) = generate_and_score(
starting_structure,
gen_name="generate_conformers_m3gnet_retrained",
gen_settings=crest_al_job.results.get_production_engine_settings(),
score_name="score_conformers_ref2",
score_settings=ref_s,
)
[13.03|14:11:33] JOB generate_conformers_m3gnet_retrained STARTED
[13.03|14:11:33] JOB generate_conformers_m3gnet_retrained RUNNING
[13.03|14:13:02] JOB generate_conformers_m3gnet_retrained FINISHED
[13.03|14:13:02] JOB generate_conformers_m3gnet_retrained SUCCESSFUL
[13.03|14:13:02] JOB score_conformers_ref2 STARTED
[13.03|14:13:02] JOB score_conformers_ref2 RUNNING
[13.03|14:13:07] JOB score_conformers_ref2 FINISHED
[13.03|14:13:07] JOB score_conformers_ref2 SUCCESSFUL
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred);
Ref_i Pred_i RMSD Ref_dE Pred_dE
0 1 0.0 0.00 0.00
1 8 0.0 0.04 0.06
2 4 0.0 0.05 0.02
3 7 0.0 0.05 0.04
... output trimmed ....
29 29 0.0 0.37 0.28
30 28 0.0 0.39 0.27
31 32 0.0 0.39 0.31
32 31 0.0 0.39 0.30
33 33 0.0 0.41 0.40
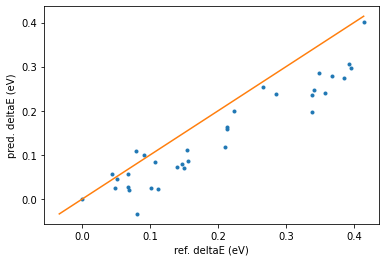
We can see a significant improvement compared to the M3GNet-UP-2022 results! However, the results are not perfect.
Generate conformers with the reference method and score with the retrained M3GNet model¶
As a second test, we can instead generate the conformers with the reference method and score them with the retrained m3gnet model.
This is quite expensive if the reference method is DFT!
if perform_expensive_tests:
generate_ref_job, molecules_ref, energies_ref, _, molecules_pred, energies_pred = (
generate_and_score(
starting_structure,
gen_name="generate_conformers_ref",
gen_settings=ref_s,
score_name="score_conformers_m3gnet_retrained",
score_settings=crest_al_job.results.get_production_engine_settings(),
)
)
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred)
plot_conformers(generate_ref_job, 3)
[13.03|14:13:09] JOB generate_conformers_ref STARTED
[13.03|14:13:09] JOB generate_conformers_ref RUNNING
[13.03|14:13:54] JOB generate_conformers_ref FINISHED
[13.03|14:13:54] JOB generate_conformers_ref SUCCESSFUL
[13.03|14:13:54] JOB score_conformers_m3gnet_retrained STARTED
... output trimmed ....
32 31 0.0 0.32 0.30
33 35 0.0 0.32 0.41
34 33 0.0 0.33 0.35
35 34 0.0 0.33 0.35
36 36 0.0 0.42 0.45
Compare the RMSD between the different conformer sets¶
The Conformer “Score” task performs single-point calculations.
If we instead change the task to “Optimize” we can see how similar the reference-method-optimized conformers are to the retrained-M3GNet-optimized conformers by comparing the RMSD.
The below is quite computationally expensive for a DFT reference engine.
if perform_expensive_tests:
opt_conformers_ref_job2 = ConformersJob(name="opt_conformers_ref2")
opt_conformers_ref_job2.settings.input.ams.Task = "Optimize"
opt_conformers_ref_job2.settings.input.ams.InputConformersSet = (
generate_conformers_m3gnet_retrained_job.results.rkfpath()
)
opt_conformers_ref_job2.settings.input.ams.InputMaxEnergy = (
5.0 # only conformers in the lowest 5.0 kcal/mol = 0.2 eV
)
opt_conformers_ref_job2.settings.input += ref_s.input
opt_conformers_ref_job2.run()
molecules_ref = opt_conformers_ref_job2.results.get_conformers()
energies_ref = opt_conformers_ref_job2.results.get_relative_energies(unit="eV")
molecules_pred = generate_conformers_m3gnet_retrained_job.results.get_conformers()
energies_pred = (
generate_conformers_m3gnet_retrained_job.results.get_relative_energies(
unit="eV"
)
)
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred)
[13.03|14:14:16] JOB opt_conformers_ref2 STARTED
[13.03|14:14:16] JOB opt_conformers_ref2 RUNNING
[13.03|14:14:37] JOB opt_conformers_ref2 FINISHED
[13.03|14:14:37] JOB opt_conformers_ref2 SUCCESSFUL
Ref_i Pred_i RMSD Ref_dE Pred_dE
... output trimmed ....
12 10 0.3 0.14 0.07
13 12 0.3 0.14 0.08
14 17 0.2 0.15 0.11
15 18 rmsd > 0.7 ang.
16 20 0.2 0.19 0.16
In the above table a few entries have “rmsd > 0.7 ang.”. This means that the reference geometry optimization causes the structure to change significantly compared to the retrained-m3gnet-optimized geometry.
In such cases it is not so meaningful to compare the relative energies between the reference and prediction, so those points are excluded from the table and from the plot.
Custom active learning loop: add newly generated conformers to training set¶
The Simple Active Learning module in AMS only works for MD simulations, so it cannot automatically add optimized conformers to the training or validation sets.
However, you can do it yourself!
The Conformers “Score” function does not store or calculate the forces. So let’s set up an AMS “Replay” job to recalculate the energies and forces to add to the training set:
replay_s = plams.Settings()
replay_s.input.ams.Task = "Replay"
replay_s.input.ams.Replay.File = (
generate_conformers_m3gnet_retrained_job.results.rkfpath()
)
replay_s.input.ams.Properties.Gradients = "Yes"
replay_s += ref_s
replay_job = plams.AMSJob(settings=replay_s, name="replay_new_conformers")
replay_job.run();
[13.03|14:14:37] JOB replay_new_conformers STARTED
[13.03|14:14:37] JOB replay_new_conformers RUNNING
[13.03|14:14:50] JOB replay_new_conformers FINISHED
[13.03|14:14:50] JOB replay_new_conformers SUCCESSFUL
Now import the data into a results importer:
ri = params.ResultsImporter.from_yaml(
crest_al_job.results.get_reference_data_directory()
)
ri.add_trajectory_singlepoints(replay_job, properties=["energy", "forces"])
yaml_dir = "data_with_conformer_singlepoints_yaml"
ri.store(yaml_dir, backup=False)
['data_with_conformer_singlepoints_yaml/job_collection.yaml',
'data_with_conformer_singlepoints_yaml/results_importer_settings.yaml',
'data_with_conformer_singlepoints_yaml/training_set.yaml',
'data_with_conformer_singlepoints_yaml/validation_set.yaml']
Then launch ParAMS:
params_job = params.ParAMSJob.from_yaml(
yaml_dir, name="params_with_conformer_singlepoints"
)
params_job.settings.input += ml_s.input.ams
params_job.settings.input.MachineLearning.LoadModel = (
crest_al_job.results.get_params_results_directory()
)
params_job.settings.input.Task = "MachineLearning"
params_job.settings.input.MachineLearning.LossCoeffs.Energy = 50
params_job.settings.input.MachineLearning.Target.Forces.Enabled = "No"
params_job.settings.input.MachineLearning.MaxEpochs = 100
params_job.run();
[13.03|14:14:52] JOB params_with_conformer_singlepoints STARTED
[13.03|14:14:52] JOB params_with_conformer_singlepoints RUNNING
[13.03|14:24:54] JOB params_with_conformer_singlepoints FINISHED
[13.03|14:24:54] JOB params_with_conformer_singlepoints SUCCESSFUL
If the job failed print the error message:
if not params_job.check():
print(params_job.get_errormsg())
Generate conformers with the new model and score with the reference method:
_, molecules_pred, energies_pred, _, molecules_ref, energies_ref = generate_and_score(
starting_structure,
gen_name="generate_conformers_m3gnet_retrained_again",
gen_settings=params_job.results.get_production_engine_settings(),
score_name="score_conformers_ref2",
score_settings=ref_s,
)
[13.03|14:24:54] JOB generate_conformers_m3gnet_retrained_again STARTED
[13.03|14:24:54] JOB generate_conformers_m3gnet_retrained_again RUNNING
[13.03|14:26:25] JOB generate_conformers_m3gnet_retrained_again FINISHED
[13.03|14:26:25] JOB generate_conformers_m3gnet_retrained_again SUCCESSFUL
[13.03|14:26:25] JOB score_conformers_ref2 STARTED
[13.03|14:26:25] Renaming job score_conformers_ref2 to score_conformers_ref2.002
[13.03|14:26:25] JOB score_conformers_ref2.002 RUNNING
[13.03|14:26:31] JOB score_conformers_ref2.002 FINISHED
[13.03|14:26:31] JOB score_conformers_ref2.002 SUCCESSFUL
And print/plot the results:
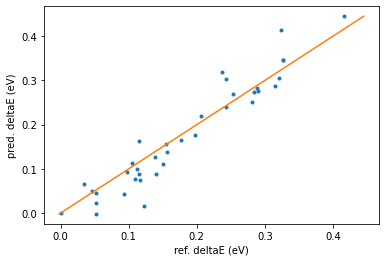
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred);
Ref_i Pred_i RMSD Ref_dE Pred_dE
0 3 0.0 0.00 0.00
1 1 0.0 0.00 -0.05
2 4 0.0 0.00 0.01
3 6 0.0 0.04 0.03
... output trimmed ....
27 28 0.0 0.30 0.31
28 27 0.0 0.32 0.30
29 29 0.0 0.39 0.34
30 30 0.0 0.40 0.41
31 31 0.0 0.51 0.46
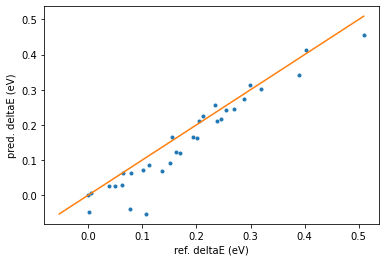
Here we see even better agreement than before.
Conclusion: By manually adding retrained-ml-optimized conformers to the training set, you can improve the conformer prediction even more. This means to do your own “active learning” outside of the Simple Active Learning module in AMS.
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# When running active learning it's usually a good idea to start off "simple" and make the system/structures gradually more complicated.
#
# For getting a model which predicts conformers accurately, we may take the following approach:
#
# * first train a potential at slightly above room temperature with NVT MD
#
# * continue training a second potential using CREST metadynamics
#
# * generate conformers with the previous model and also train to those
#
# ## Initial imports
import scm.plams as plams
import scm.params as params
from scm.simple_active_learning import SimpleActiveLearningJob
import matplotlib.pyplot as plt
import os
import numpy as np
from typing import List
from scm.conformers import ConformersJob
from scm.conformers.plams.plot import plot_conformers
plams.init()
# ## Create the initial structure
molecule = plams.from_smiles("OC(CC1c2ccccc2Sc2ccccc21)CN1CCCC1", forcefield="uff")
molecule.delete_all_bonds()
for at in molecule:
at.properties = plams.Settings()
plams.plot_molecule(molecule)
starting_structure = molecule
# ## Reference engine settings
fast_ref_s = plams.Settings()
fast_ref_s.input.DFTB.Model = "GFN1-xTB"
slow_ref_s = plams.Settings()
slow_ref_s.input.ADF.Basis.Type = "TZP"
slow_ref_s.input.ADF.Basis.Core = "None"
slow_ref_s.input.ADF.XC.Hybrid = "B3LYP"
slow_ref_s.input.ADF.XC.DISPERSION = "GRIMME3 BJDAMP"
# Change to slow_ref_s to train to B3LYP-D3(BJ) instead:
ref_s = fast_ref_s.copy()
# ref_s = slow_ref_s.copy()
perform_expensive_tests = ref_s == fast_ref_s
# ## Problem statement: Generate a few conformers with M3GNet-UP-2022 and Score with reference method
m3gnet_up_s = plams.Settings()
m3gnet_up_s.input.MLPotential.Model = "M3GNet-UP-2022"
generate_conformers_m3gnet_up_job = ConformersJob(
name="generate_conformers_m3gnet_up", molecule=starting_structure
)
generate_conformers_m3gnet_up_job.settings.input.ams.Task = "Generate"
generate_conformers_m3gnet_up_job.settings.input.ams.Generator.Method = "RDKit"
generate_conformers_m3gnet_up_job.settings.input.ams.Generator.RDKit.InitialNConformers = 40
# generate_conformers_m3gnet_up_job.settings += crest_al_job.results.get_production_engine_settings()
generate_conformers_m3gnet_up_job.settings += m3gnet_up_s
# generate_conformers_m3gnet_up_job.settings.runscript.nproc = 1 # run in serial, useful if you run out of memory when running M3GNet on the GPU
generate_conformers_m3gnet_up_job.run();
# Show the 4 most stable conformers with M3GNet-UP-2022:
plot_conformers(generate_conformers_m3gnet_up_job, 4, unit="eV");
# Score the generated conformers with the reference method:
score_conformers_ref_job = ConformersJob(name="score_conformers_ref")
score_conformers_ref_job.settings.input.ams.Task = "Score"
score_conformers_ref_job.settings.input.ams.InputConformersSet = (
generate_conformers_m3gnet_up_job.results.rkfpath()
)
score_conformers_ref_job.settings.input += ref_s.input
score_conformers_ref_job.run();
plot_conformers(score_conformers_ref_job, 4, unit="eV");
# Here we can see that the ordering of conformers at the reference level is completely different compared to M3GNet-UP-2022.
#
# The Score job reorders the conformers. To compare the energies more precisely, we can use the minimum pairwise RMSD (which should be 0) to identify how the order of conformers change:
def get_pairwise_rmsds(
molecules_ref: List[plams.Molecule], molecules_pred: List[plams.Molecule]
) -> np.ndarray:
"""Returns a len(molecules_ref)*len(molecules_pred) numpy array with pairwise rmsds (in angstrom) between the structures"""
rmsds = np.zeros((len(molecules_ref), len(molecules_pred)))
for i, ref_mol in enumerate(molecules_ref):
for j, pred_mol in enumerate(molecules_pred):
rmsds[i, j] = plams.Molecule.rmsd(ref_mol, pred_mol)
return rmsds
def get_reordering(rmsds: np.ndarray) -> np.ndarray:
"""
rmsds: numpy array with shape len(molecules_ref)*len(molecules_pred)
Returns a len(molecules_ref) integer numpy array.
The first element is the index (0-based) in molecules_pred corresponding to the first reference molecule, etc.
"""
rmsds = get_pairwise_rmsds(molecules_ref, molecules_pred)
reordering = np.argmin(rmsds, axis=1)
return reordering
def print_reordering_table(
molecules_ref, molecules_pred, energies_ref, energies_pred, ax=None
):
"""This functions prints the reordering table including relative energies. It also plots the predicted relative energies versus the reference relative energies"""
print(f"Ref_i Pred_i RMSD Ref_dE Pred_dE")
x, y = [], []
rmsds = get_pairwise_rmsds(molecules_ref, molecules_pred)
reordering = get_reordering(rmsds)
rmsd_threshold = 0.7 # angstrom, for printing/plotting points
for ref_i, pred_i in enumerate(reordering):
rmsd = rmsds[ref_i, pred_i]
ref_relative_e = energies_ref[ref_i]
pred_relative_e = energies_pred[pred_i] - energies_pred[reordering[0]]
if rmsd <= rmsd_threshold:
print(
f"{ref_i:6d} {pred_i:6d} {rmsd:4.1f} {ref_relative_e:7.2f} {pred_relative_e:7.2f}"
)
x.append(ref_relative_e)
y.append(pred_relative_e)
else:
print(f"{ref_i:6d} {pred_i:6d} rmsd > {rmsd_threshold:.1f} ang.")
if ax is None:
_, ax = plt.subplots()
m, M = np.min([np.min(x), np.min(y)]), np.max([np.max(x), np.max(y)])
ax.plot(x, y, ".")
ax.plot([m, M], [m, M], "-")
ax.set_xlabel("ref. deltaE (eV)")
ax.set_ylabel("pred. deltaE (eV)")
return ax
# In the below table the energies are given with respect to the structure that had the lowest reference energy. This is thus different compared to the first image above, where the predicted (m3gnet-up-2022) relative energies were given to predicted lowest energy conformer.
molecules_ref = score_conformers_ref_job.results.get_conformers()
energies_ref = score_conformers_ref_job.results.get_relative_energies(unit="eV")
molecules_pred = generate_conformers_m3gnet_up_job.results.get_conformers()
energies_pred = generate_conformers_m3gnet_up_job.results.get_relative_energies(
unit="ev"
)
ax = print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred)
ax.set_title("M3GNet-UP-2022 relative to reference method");
ax.figure.savefig("picture1.png")
# Here we see that there is no correlation between the relative stabilities calculated with M3GNet-UP-2022 and the reference method.
#
# Example: The most stable conformer with the reference method (Ref_i=0) corresponds to the tenth most stable conformer with M3GNet-UP-2022 (Pred_i=10)
#
# Example 2: The second most stable conformer with the reference method (Ref_i=1) should be 0.08 eV *less* stable than the ref_i=0 conformer, but with M3GNet-UP-2022 method it is actually 0.01 eV *more* stable!
# ## Optimize a few conformers for initial reference data
#
# Conformers are local minima on the potential energy surface. The Active Learning MD will sample off-equilibrium structures. So let's make sure that there are at least some local minima in the training set by optimizing some of the generated conformers.
#
# Here, we loop over the conformers and run GeometryOptimization jobs. This produces output files in the normal AMS format that can easily be imported into ParAMS
max_N = min(6, len(molecules_pred)) # at most 6 optimizations
opt_s = plams.Settings()
opt_s.input.ams.Task = "GeometryOptimization"
opt_s.input.ams.GeometryOptimization.Convergence.Quality = "Basic"
opt_jobs = [
plams.AMSJob(settings=opt_s + ref_s, name=f"opt_{i}", molecule=mol)
for i, mol in enumerate(molecules_pred[:max_N])
]
for opt_job in opt_jobs:
opt_job.run()
# Now import the data into a ParAMS results importer and store in the directory ``my_initial_reference_data``:
yaml_dir = os.path.abspath("my_initial_reference_data")
ri = params.ResultsImporter(
settings={"units": {"energy": "eV", "forces": "eV/angstrom"}}
)
for opt_job in opt_jobs:
ri.add_singlejob(opt_job, properties=["energy", "forces"], task="SinglePoint")
ri.store(yaml_dir, backup=False)
# ## Simple Active Learning setup
#
# ### Molecular dynamics settings (temperature ramp)
nvt_md_s = plams.AMSNVTJob(
nsteps=20000,
timestep=0.5,
temperature=(270, 350, 350),
tau=100,
thermostat="Berendsen",
).settings
# ### ParAMS machine learning settings
ml_s = plams.Settings()
ml_s.input.ams.MachineLearning.Backend = "M3GNet"
ml_s.input.ams.MachineLearning.CommitteeSize = 1
ml_s.input.ams.MachineLearning.M3GNet.Model = "UniversalPotential"
ml_s.input.ams.MachineLearning.MaxEpochs = 200
# ### Active learning settings
#
# Conformer search of a single molecule is quite simple, so we can expect the ML method to perform quite well. We therefore decrease the success criteria thresholds a bit compared to the default values, to ensure that we get accurate results.
#
# Since we will immediately continue with another active learning loop, we disable the "RerunSimulation" as we are not interested in the MD simulation per se.
al_s = plams.Settings()
al_s.input.ams.ActiveLearning.Steps.Type = "Geometric"
al_s.input.ams.ActiveLearning.Steps.Geometric.Start = 10
al_s.input.ams.ActiveLearning.Steps.Geometric.NumSteps = 8
al_s.input.ams.ActiveLearning.InitialReferenceData.Load.Directory = yaml_dir
al_s.input.ams.ActiveLearning.InitialReferenceData.Generate.M3GNetShortMD.Enabled = (
"Yes"
)
al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.002
al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = 0.30
al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
# ### Initial structure
#
# Let's start with the lowest-energy conformer according to the reference method:
starting_structure_al = molecules_ref[0]
# The bonds are not used, so delete them to make the input file less confusing:
starting_structure_al.delete_all_bonds()
# ### Complete job
settings = ref_s + nvt_md_s + ml_s + al_s
job = SimpleActiveLearningJob(
settings=settings, molecule=starting_structure_al, name="sal"
)
print(job.get_input())
# ### Run the simple active learning job
job.run(watch=True);
# ### Final structure from MD simulation
#
new_structure = job.results.get_main_molecule()
plams.plot_molecule(new_structure, rotation="-5x,5y,0z")
# ## Second active learning job: CREST metadynamics
#
# Here we set Steps.Type = "Linear" to run reference calculations every 2000 MD steps.
nsteps = 20000
crest_md_s = plams.Settings()
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.Height = 0.138
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.NGaussiansMax = 50
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.NSteps = 200
crest_md_s.input.ams.MolecularDynamics.CRESTMTD.Width = 0.62
crest_complete_md_s = plams.AMSMDJob(
molecule=new_structure,
nsteps=nsteps,
settings=crest_md_s,
tau=10, # small time constant
thermostat="NHC",
temperature=300,
timestep=0.5,
samplingfreq=20,
).settings
# job = SimpleActiveLearningJob.load_external(plams.config.default_jobmanager.workdir + "/sal.002")
crest_ml_s = ml_s.copy()
crest_ml_s.input.ams.MachineLearning.LoadModel = (
job.results.get_params_results_directory()
)
crest_ml_s.input.ams.MachineLearning.Target.Forces.MAE = 0.04
crest_ml_s.input.ams.MachineLearning.MaxEpochs = 200
crest_al_s = plams.Settings()
crest_al_s.input.ams.ActiveLearning.Steps.Type = "Linear"
crest_al_s.input.ams.ActiveLearning.Steps.Linear.Start = 500
crest_al_s.input.ams.ActiveLearning.Steps.Linear.StepSize = 2000
crest_al_s.input.ams.ActiveLearning.InitialReferenceData.Load.FromPreviousModel = "Yes"
crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Relative = 0.002
crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Total = 0.010
# because we do not set Normalization, the above Energy criteria are energies per atom
# crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Energy.Normalization =
crest_al_s.input.ams.ActiveLearning.SuccessCriteria.Forces.MaxDeviationForZeroForce = (
0.30
)
crest_al_s.input.ams.ActiveLearning.AtEnd.RerunSimulation = "No"
crest_al_s.input.ams.ActiveLearning.MaxReferenceCalculationsPerAttempt = 3
crest_al_job = SimpleActiveLearningJob(
name="crest_al",
settings=ref_s + crest_complete_md_s + crest_ml_s + crest_al_s,
molecule=new_structure,
)
crest_al_job.run(watch=True);
# crest_al_job = SimpleActiveLearningJob.load_external("plams_workdir.003/crest_al")
new_retrained_model_settings = (
crest_al_job.results.get_params_job().results.get_production_engine_settings()
)
# ## Generate conformers with the retrained M3GNet model and score with reference method
def generate_and_score(
molecule: plams.Molecule,
gen_name: str,
gen_settings: plams.Settings,
score_name: str,
score_settings: plams.Settings,
):
generate_job = ConformersJob(name=gen_name, molecule=molecule)
generate_job.settings.input.ams.Task = "Generate"
generate_job.settings.input.ams.Generator.Method = "RDKit"
generate_job.settings.input.ams.Generator.RDKit.InitialNConformers = 40
generate_job.settings.input += gen_settings.input
generate_job.run()
score_job = ConformersJob(name=score_name)
score_job.settings.input.ams.Task = "Score"
score_job.settings.input.ams.InputConformersSet = generate_job.results.rkfpath()
score_job.settings.input += score_settings.input
score_job.run()
molecules_gen = generate_job.results.get_conformers()
energies_gen = generate_job.results.get_relative_energies(unit="eV")
molecules_score = score_job.results.get_conformers()
energies_score = score_job.results.get_relative_energies(unit="ev")
return (
generate_job,
molecules_gen,
energies_gen,
score_job,
molecules_score,
energies_score,
)
(
generate_conformers_m3gnet_retrained_job,
molecules_pred,
energies_pred,
_,
molecules_ref,
energies_ref,
) = generate_and_score(
starting_structure,
gen_name="generate_conformers_m3gnet_retrained",
gen_settings=crest_al_job.results.get_production_engine_settings(),
score_name="score_conformers_ref2",
score_settings=ref_s,
)
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred);
# We can see a significant improvement compared to the M3GNet-UP-2022 results! However, the results are not perfect.
# ## Generate conformers with the reference method and score with the retrained M3GNet model
#
# As a second test, we can instead generate the conformers with the reference method and score them with the retrained m3gnet model.
#
# This is quite expensive if the reference method is DFT!
if perform_expensive_tests:
generate_ref_job, molecules_ref, energies_ref, _, molecules_pred, energies_pred = (
generate_and_score(
starting_structure,
gen_name="generate_conformers_ref",
gen_settings=ref_s,
score_name="score_conformers_m3gnet_retrained",
score_settings=crest_al_job.results.get_production_engine_settings(),
)
)
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred)
plot_conformers(generate_ref_job, 3)
# ## Compare the RMSD between the different conformer sets
#
# The Conformer "Score" task performs single-point calculations.
#
# If we instead change the task to "Optimize" we can see how similar the reference-method-optimized conformers are to the retrained-M3GNet-optimized conformers by comparing the RMSD.
#
# The below is quite computationally expensive for a DFT reference engine.
if perform_expensive_tests:
opt_conformers_ref_job2 = ConformersJob(name="opt_conformers_ref2")
opt_conformers_ref_job2.settings.input.ams.Task = "Optimize"
opt_conformers_ref_job2.settings.input.ams.InputConformersSet = (
generate_conformers_m3gnet_retrained_job.results.rkfpath()
)
opt_conformers_ref_job2.settings.input.ams.InputMaxEnergy = (
5.0 # only conformers in the lowest 5.0 kcal/mol = 0.2 eV
)
opt_conformers_ref_job2.settings.input += ref_s.input
opt_conformers_ref_job2.run()
molecules_ref = opt_conformers_ref_job2.results.get_conformers()
energies_ref = opt_conformers_ref_job2.results.get_relative_energies(unit="eV")
molecules_pred = generate_conformers_m3gnet_retrained_job.results.get_conformers()
energies_pred = (
generate_conformers_m3gnet_retrained_job.results.get_relative_energies(
unit="eV"
)
)
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred)
# In the above table a few entries have "rmsd > 0.7 ang.". This means that the reference geometry optimization causes the structure to change significantly compared to the retrained-m3gnet-optimized geometry.
#
# In such cases it is not so meaningful to compare the relative energies between the reference and prediction, so those points are excluded from the table and from the plot.
# ## Custom active learning loop: add newly generated conformers to training set
#
# The Simple Active Learning module in AMS only works for MD simulations, so it cannot automatically add optimized conformers to the training or validation sets.
#
# However, you can do it yourself!
#
# The Conformers "Score" function does not store or calculate the forces. So let's set up an AMS "Replay" job to recalculate the energies and forces to add to the training set:
replay_s = plams.Settings()
replay_s.input.ams.Task = "Replay"
replay_s.input.ams.Replay.File = (
generate_conformers_m3gnet_retrained_job.results.rkfpath()
)
replay_s.input.ams.Properties.Gradients = "Yes"
replay_s += ref_s
replay_job = plams.AMSJob(settings=replay_s, name="replay_new_conformers")
replay_job.run();
# Now import the data into a results importer:
ri = params.ResultsImporter.from_yaml(
crest_al_job.results.get_reference_data_directory()
)
ri.add_trajectory_singlepoints(replay_job, properties=["energy", "forces"])
yaml_dir = "data_with_conformer_singlepoints_yaml"
ri.store(yaml_dir, backup=False)
# Then launch ParAMS:
params_job = params.ParAMSJob.from_yaml(
yaml_dir, name="params_with_conformer_singlepoints"
)
params_job.settings.input += ml_s.input.ams
params_job.settings.input.MachineLearning.LoadModel = (
crest_al_job.results.get_params_results_directory()
)
params_job.settings.input.Task = "MachineLearning"
params_job.settings.input.MachineLearning.LossCoeffs.Energy = 50
params_job.settings.input.MachineLearning.Target.Forces.Enabled = "No"
params_job.settings.input.MachineLearning.MaxEpochs = 100
params_job.run();
# If the job failed print the error message:
if not params_job.check():
print(params_job.get_errormsg())
# Generate conformers with the new model and score with the reference method:
_, molecules_pred, energies_pred, _, molecules_ref, energies_ref = generate_and_score(
starting_structure,
gen_name="generate_conformers_m3gnet_retrained_again",
gen_settings=params_job.results.get_production_engine_settings(),
score_name="score_conformers_ref2",
score_settings=ref_s,
)
# And print/plot the results:
print_reordering_table(molecules_ref, molecules_pred, energies_ref, energies_pred);
# Here we see even better agreement than before.
#
# **Conclusion**: By manually adding retrained-ml-optimized conformers to the training set, you can improve the conformer prediction even more. This means to do your own "active learning" outside of the Simple Active Learning module in AMS.