Active Learning: Ru/H Part 1, Initial Reference Scans¶
Introducing the Ru/H case study¶
Trained model: M3GNet, starting from the Universal Potential (UP)
Reference method: PBE-D3(BJ) with engine Quantum ESPRESSO
System: H atoms depositing onto Ru surfaces
Problem: M3GNet-UP-2022 is not very reliable for high-temperature surface chemistry.
Solution: Retraining the model gives better agreement.
Expected duration: This example takes several days to run on a modern compute node.
This is a very thorough example which shows how to
construct initial reference data using PES Scans like volume scans, cartesian coordinate scans, and bond scans as well as MD simulations
training an initial model to the reference data before the active learning loop
running an active learning loop with the molecule gun
Important
The common_ru_h.py file contains a variable TESTING_MODE.
Set TESTING_MODE = True to not use DFT reference calculations but instead a custom-trained M3GNet model for the reference calculations. This will let you run through the workflow quickly without running any expensive DFT reference calculations.
Here in part 1, we start the Ru/H active-learning case study by generating initial reference data from a bulk Ru lattice optimization, a volume scan, and an H2 bond scan.
Downloads: Notebook | Script ?
Additional files required by this example:
common_ru_h.py
Requires: AMS2026 or later
Required AMS Packages: m3gnet
Related examples
Related tutorials
Related documentation
Initial imports¶
import scm.plams as plams
from scm.params import ResultsImporter
from scm.plams import Settings, AMSJob, log, Molecule
# common_ru_h.py must exist in the current working directory
from common_ru_h import (
rotation,
dft_settings,
QEKPointsConfig,
lattice_optimization_settings,
plot_pesscan,
check_installation,
)
# register dependencies for AMSjobs, to support submitting this notebook directly to a cluster in AMS2025+
# dependency: {} common_ru_h.py
check_installation()
Current AMS version: 2024.102
05-31 11:12:18 m3gnet is installed: M3GNet ML Backend v[0.2.4] - build:0 [06668e0a45ce742d8f66ff23484b8a1e]
05-31 11:12:18 qe is installed: Quantum ESPRESSO (AMSPIPE) v[7.1] - build:115 [777d72eb480fe4d632a003cc62e9c1cb]
Initialize PLAMS working directory¶
plams.init()
PLAMS working folder: /path/to/plams_workdir
Bulk structure: hcp Ru¶
initial_bulk = plams.Molecule()
a = 2.7 # hexagonal lattice parameter, angstrom
c = 4.2768 # hexagonal lattice parameter, angstrom
initial_bulk.add_atom(plams.Atom(symbol="Ru", coords=(0.0, 0.0, 0.0)))
initial_bulk.add_atom(plams.Atom(symbol="Ru", coords=(0.0, a / 3**0.5, c / 2)))
initial_bulk.lattice = [[a, 0, 0], [-a / 2, a * 3**0.5 / 2, 0], [0, 0, c]]
log("Initial structure")
log(initial_bulk)
[31.05|11:12:18] Initial structure
[31.05|11:12:18] Atoms:
1 Ru 0.000000 0.000000 0.000000
2 Ru 0.000000 1.558846 2.138400
Lattice:
2.7000000000 0.0000000000 0.0000000000
-1.3500000000 2.3382685902 0.0000000000
0.0000000000 0.0000000000 4.2768000000
plams.plot_molecule(initial_bulk, rotation=rotation)

Lattice optimization of bulk Ru with DFT¶
lattopt_job = plams.AMSJob(
settings=dft_settings(QEKPointsConfig(11, 11, 11))
+ lattice_optimization_settings(),
name="hcp_lattopt_Ru_dft",
molecule=initial_bulk,
)
lattopt_job.run();
[31.05|11:12:19] JOB hcp_lattopt_Ru_dft STARTED
[31.05|11:12:19] JOB hcp_lattopt_Ru_dft RUNNING
[31.05|11:12:44] JOB hcp_lattopt_Ru_dft FINISHED
[31.05|11:12:45] JOB hcp_lattopt_Ru_dft SUCCESSFUL
optimized_bulk: Molecule = lattopt_job.results.get_main_molecule() # type: ignore
log(optimized_bulk)
log(f"Density: {optimized_bulk.get_density():.2f} kg/m^3")
[31.05|11:12:45] Atoms:
1 Ru 0.000000 0.000000 0.000000
2 Ru -0.000000 1.561640 2.136759
Lattice:
2.7048396424 0.0000000000 0.0000000000
-1.3524198212 2.3424598435 0.0000000000
0.0000000000 0.0000000000 4.2735187675
[31.05|11:12:45] Density: 12396.59 kg/m^3
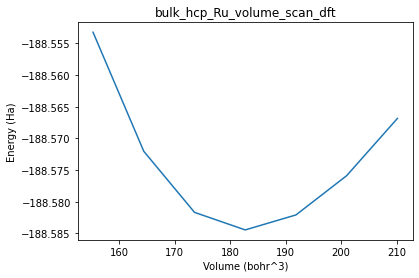
Volume scan of bulk Ru with DFT¶
from common_ru_h import (
dft_settings,
QEKPointsConfig,
pesscan_settings,
CellVolumeScalingRangeScanCoordinate,
)
s = dft_settings(QEKPointsConfig(11, 11, 11))
s += pesscan_settings([CellVolumeScalingRangeScanCoordinate(0.85, 1.15)], n_points=7)
volume_scan_job = AMSJob(
settings=s,
molecule=optimized_bulk,
name="bulk_hcp_Ru_volume_scan_dft",
)
volume_scan_job.run();
[31.05|11:12:45] JOB bulk_hcp_Ru_volume_scan_dft STARTED
[31.05|11:12:45] JOB bulk_hcp_Ru_volume_scan_dft RUNNING
[31.05|11:14:17] JOB bulk_hcp_Ru_volume_scan_dft FINISHED
[31.05|11:14:18] JOB bulk_hcp_Ru_volume_scan_dft SUCCESSFUL
plot_pesscan(volume_scan_job);
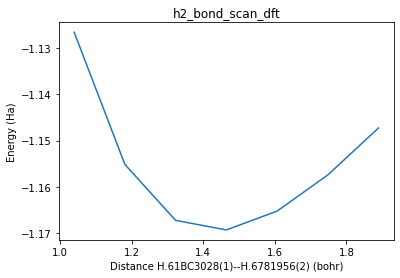
Bond scan of H2 with DFT¶
h2_mol = plams.from_smiles("[HH]")
h2_mol.lattice = [[5, 0, 0], [0, 5, 0], [0, 0, 5]]
plams.plot_molecule(h2_mol, rotation=rotation)

from common_ru_h import (
dft_settings,
QEKPointsConfig,
pesscan_settings,
DistanceScanCoordinate,
)
scan_coordinate = DistanceScanCoordinate(atom1=1, atom2=2, start=0.55, end=1.0)
s = dft_settings(QEKPointsConfig(1, 1, 1))
s += pesscan_settings([scan_coordinate], n_points=7)
h2_bond_scan_job = AMSJob(settings=s, molecule=h2_mol, name="h2_bond_scan_dft")
h2_bond_scan_job.run();
[31.05|11:14:18] JOB h2_bond_scan_dft STARTED
[31.05|11:14:18] JOB h2_bond_scan_dft RUNNING
[31.05|11:15:19] JOB h2_bond_scan_dft FINISHED
[31.05|11:15:19] Job h2_bond_scan_dft reported warnings. Please check the output
[31.05|11:15:19] JOB h2_bond_scan_dft SUCCESSFUL
plot_pesscan(h2_bond_scan_job);
Store results¶
ri = ResultsImporter.from_ase()
properties = ["energy", "forces"]
ri.add_pesscan_singlepoints(volume_scan_job, properties=properties)
ri.add_pesscan_singlepoints(h2_bond_scan_job, properties=properties)
ri.add_singlejob(lattopt_job, task="SinglePoint", properties=properties)
# Also add as PES Scans - these will not be used during training but
# will plot the energy-volume curve and bond-scan curve at the end
# of the training
ri.add_singlejob(volume_scan_job, task="PESScan", properties=["pes"])
ri.add_singlejob(h2_bond_scan_job, task="PESScan", properties=["pes"])
ri.store("reference_data_1")
['reference_data_1/job_collection.yaml',
'reference_data_1/results_importer_settings.yaml',
'reference_data_1/training_set.yaml']
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial imports
import scm.plams as plams
from scm.params import ResultsImporter
from scm.plams import Settings, AMSJob, log, Molecule
# common_ru_h.py must exist in the current working directory
from common_ru_h import (
rotation,
dft_settings,
QEKPointsConfig,
lattice_optimization_settings,
plot_pesscan,
check_installation,
)
# register dependencies for AMSjobs, to support submitting this notebook directly to a cluster in AMS2025+
# dependency: {} common_ru_h.py
check_installation()
# ## Initialize PLAMS working directory
plams.init()
# ## Bulk structure: hcp Ru
initial_bulk = plams.Molecule()
a = 2.7 # hexagonal lattice parameter, angstrom
c = 4.2768 # hexagonal lattice parameter, angstrom
initial_bulk.add_atom(plams.Atom(symbol="Ru", coords=(0.0, 0.0, 0.0)))
initial_bulk.add_atom(plams.Atom(symbol="Ru", coords=(0.0, a / 3**0.5, c / 2)))
initial_bulk.lattice = [[a, 0, 0], [-a / 2, a * 3**0.5 / 2, 0], [0, 0, c]]
log("Initial structure")
log(initial_bulk)
plams.plot_molecule(initial_bulk, rotation=rotation)
# ## Lattice optimization of bulk Ru with DFT
lattopt_job = plams.AMSJob(
settings=dft_settings(QEKPointsConfig(11, 11, 11)) + lattice_optimization_settings(),
name="hcp_lattopt_Ru_dft",
molecule=initial_bulk,
)
lattopt_job.run()
optimized_bulk: Molecule = lattopt_job.results.get_main_molecule() # type: ignore
log(optimized_bulk)
log(f"Density: {optimized_bulk.get_density():.2f} kg/m^3")
# ## Volume scan of bulk Ru with DFT
from common_ru_h import (
dft_settings,
QEKPointsConfig,
pesscan_settings,
CellVolumeScalingRangeScanCoordinate,
)
s = dft_settings(QEKPointsConfig(11, 11, 11))
s += pesscan_settings([CellVolumeScalingRangeScanCoordinate(0.85, 1.15)], n_points=7)
volume_scan_job = AMSJob(
settings=s,
molecule=optimized_bulk,
name="bulk_hcp_Ru_volume_scan_dft",
)
volume_scan_job.run()
plot_pesscan(volume_scan_job)
# ## Bond scan of H2 with DFT
h2_mol = plams.from_smiles("[HH]")
h2_mol.lattice = [[5, 0, 0], [0, 5, 0], [0, 0, 5]]
plams.plot_molecule(h2_mol, rotation=rotation)
from common_ru_h import (
dft_settings,
QEKPointsConfig,
pesscan_settings,
DistanceScanCoordinate,
)
scan_coordinate = DistanceScanCoordinate(atom1=1, atom2=2, start=0.55, end=1.0)
s = dft_settings(QEKPointsConfig(1, 1, 1))
s += pesscan_settings([scan_coordinate], n_points=7)
h2_bond_scan_job = AMSJob(settings=s, molecule=h2_mol, name="h2_bond_scan_dft")
h2_bond_scan_job.run()
plot_pesscan(h2_bond_scan_job)
# ## Store results
ri = ResultsImporter.from_ase()
properties = ["energy", "forces"]
ri.add_pesscan_singlepoints(volume_scan_job, properties=properties)
ri.add_pesscan_singlepoints(h2_bond_scan_job, properties=properties)
ri.add_singlejob(lattopt_job, task="SinglePoint", properties=properties)
# Also add as PES Scans - these will not be used during training but
# will plot the energy-volume curve and bond-scan curve at the end
# of the training
ri.add_singlejob(volume_scan_job, task="PESScan", properties=["pes"])
ri.add_singlejob(h2_bond_scan_job, task="PESScan", properties=["pes"])
ri.store("reference_data_1")