Conformer Generation for Multiple Molecules¶
Apply AMS conformer generation to a system with more than one molecule.
Conformer generation for multiple molecules¶
Lets see how two alanine molecules orient themselves using CREST conformer generation. To do this we will constrain the system in a spherical region using the SphericalWall constraint. We start by setting up a system of two alanine molecules in a relatively small space.
Initial imports¶
import sys
import random
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import scm.plams as plams
from scm.base import ChemicalSystem
from scm.conformers import ConformersJob
from scm.utils.conversions import chemsys_to_plams_molecule, plams_molecule_to_chemsys
Alanine dimer¶
from scm.plams import preoptimize
smiles = "CC(N)C(=O)O.CC(N)C(=O)O"
dimer = ChemicalSystem.from_smiles(smiles) # alternatively you may use packmol to build the dimer
dimer.translate(-dimer.center_of_mass())
Optimize the dimer structure prior to the conformer search.
s = plams.Settings()
s.input.GFNFF = plams.Settings()
s.input.ams.Task = "GeometryOptimization"
s.input.ams.GeometryOptimization.Convergence.Quality = "VeryGood"
s.input.ams.GeometryOptimization.Maxiterations = 1300
s.input.ams.GeometryOptimization.Method = "Quasi-Newton"
job = plams.AMSJob(molecule=dimer, settings=s)
job.run()
dimer = job.results.get_main_system()
plams.view(dimer, height=300, width=300, direction="along_pca3")
[09.04|12:12:09] JOB plamsjob STARTED
[09.04|12:12:09] JOB plamsjob RUNNING
[09.04|12:12:11] JOB plamsjob FINISHED
[09.04|12:12:11] JOB plamsjob SUCCESSFUL
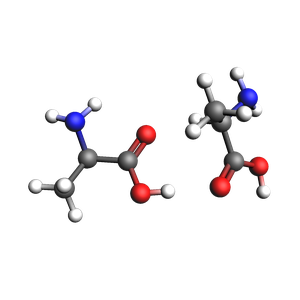
Calculation setup¶
To determine the radius of the SphericalWall we measure the size of the initial dimer.
dists = dimer.distance_matrix()
max_dist = np.max(dists)
diameter = 1.33 * max_dist
radius = diameter / 2
print(f"Largest distance between atoms: {max_dist:.3f} ang.")
print(f"Radius: {radius:.3f} ang.")
Largest distance between atoms: 8.779 ang.
Radius: 5.838 ang.
Now we can set up the Crest conformer generation job, with the appropriate spherical wall constraining the molecules close together.
# This seed will used for the starting MD velocities in conformers generation
# Due to numerical aspects it does not guarantee full reproducibility.
seed = random.randint(1, 10000000)
print(f"Seed used for stochastic aspects is {seed}.")
Seed used for stochastic aspects is 3330407.
settings = plams.Settings()
settings.input.ams.EngineAddons.WallPotential.Enabled = "Yes"
settings.input.ams.EngineAddons.WallPotential.Radius = radius
settings.input.ams.Generator.Method = "CREST"
settings.input.ams.Output.KeepWorkDir = "Yes"
settings.input.ams.GeometryOptimization.MaxConvergenceTime = "High"
settings.input.ams.Generator.CREST.NCycles = 3 # at most 3 CREST cycles for this demo
settings.input.ams.Generator.RNGSeed = seed
settings.input.ams.Generator.CREST.NMolDynStepsFactor = 3
settings.input.GFNFF = plams.Settings()
Run the conformers job¶
Now we can run the conformer generation job. This job will run somewhere between 30 minutes and 1 hour.
job = ConformersJob(molecule=chemsys_to_plams_molecule(dimer), settings=settings)
job.run();
# ConformersJob.load_external("plams_workdir/conformers/conformers.rkf") # load from disk instead of running the job
[09.04|12:12:13] JOB conformers STARTED
[09.04|12:12:13] JOB conformers RUNNING
[09.04|12:25:10] JOB conformers FINISHED
[09.04|12:25:10] JOB conformers SUCCESSFUL
Now, remove the wall and reoptimize
if "EngineAddons" in settings.input.ams:
del settings.input.ams.EngineAddons
settings.input.ams.Task = "Optimize"
settings.input.ams.InputConformersSet = job.results.rkfpath()
opt_job = ConformersJob(settings=settings)
opt_job.run();
[09.04|12:25:10] JOB conformers STARTED
[09.04|12:25:10] Renaming job conformers to conformers.002
[09.04|12:25:10] JOB conformers.002 RUNNING
[09.04|12:26:43] JOB conformers.002 FINISHED
[09.04|12:26:43] JOB conformers.002 SUCCESSFUL
rkf = opt_job.results.rkfpath()
print(f"Conformers stored in {rkf}")
Conformers stored in /Users/ormrodmorley/Documents/code/ams/amshome/userdoc/PythonExamples/conformers-multiple-molecules/plams_workdir/conformers.002/conformers.rkf
Results¶
relative_energies = opt_job.results.get_relative_energies("kcal/mol")
print(f"Total number of conformers: {len(relative_energies)}")
conformer_table = pd.DataFrame(
{
"Conformer Id": np.arange(1, len(relative_energies) + 1),
"Rel. E (kcal/mol)": relative_energies,
"Population at 298 K": opt_job.results.get_boltzmann_distribution(298),
}
)
conformer_table.head(20)
Total number of conformers: 1678
Conformer Id | Rel. E (kcal/mol) | Population at 298 K | |
|---|---|---|---|
0 | 1 | 0.000000 | 0.079996 |
1 | 2 | 0.007065 | 0.079047 |
2 | 3 | 0.011192 | 0.078498 |
3 | 4 | 0.071612 | 0.070884 |
4 | 5 | 0.103091 | 0.067215 |
5 | 6 | 0.205166 | 0.056572 |
6 | 7 | 0.227356 | 0.054492 |
7 | 8 | 0.305097 | 0.047788 |
8 | 9 | 0.314653 | 0.047023 |
9 | 10 | 0.348506 | 0.044410 |
10 | 11 | 0.437493 | 0.038214 |
11 | 12 | 0.470471 | 0.036144 |
12 | 13 | 0.494662 | 0.034697 |
13 | 14 | 0.519705 | 0.033261 |
14 | 15 | 0.934194 | 0.016518 |
15 | 16 | 0.948115 | 0.016134 |
16 | 17 | 0.967742 | 0.015608 |
17 | 18 | 1.094571 | 0.012599 |
18 | 19 | 1.370615 | 0.007905 |
19 | 20 | 1.374833 | 0.007849 |
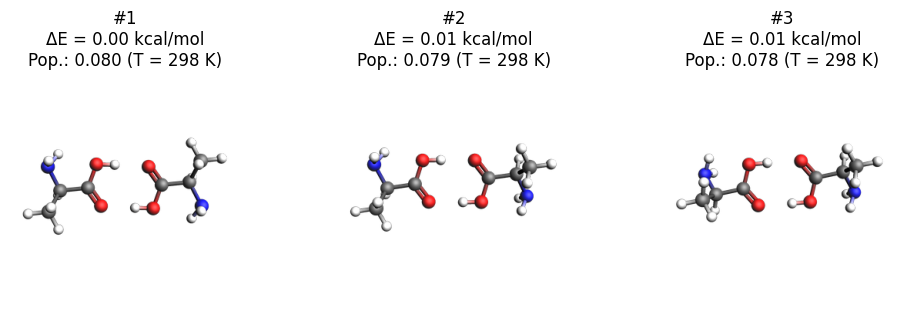
Here we plot the three lowest-energy conformers.
from scm.conformers.plams.plot import plot_conformers
axes = plot_conformers(opt_job, width=300, height=300, direction="along_pca3")
axes;
You can also open the conformers in AMSmovie to browse all conformers 500+ conformers:
!amsmovie "{opt_job.results.rkfpath()}"
Filtering out duplicates¶
The conformerset attribute holds information about the full set of conformers.
conformerset = opt_job.results.conformerset
print("Number of conforrmers: ", len(conformerset))
Number of conforrmers: 1678
This conformerset does not contain any duplicates. However, there is no single definition of the term duplicates when applied to conformers. By default, duplicates are defined based on energy and rotational constants, the latter quantifying the 3D shape of the system. The filtering method uses thresholds for energy and rotational constants, which have been thoroughly tested for (mostly small) single molecules systems. If the user feels that the stored conformers are too similar, these thresholds can of course be changed. Below we refilter the stored conformerset using more lenient thresholds (systems with greater differences will be considered duplicates).
s = plams.Settings()
s.input.ams.Task = "Filter"
s.input.ams.InputConformersSet = rkf
s.input.ams.Equivalence.CREST.EnergyThreshold = 0.2 # default is 0.05
s.input.ams.Equivalence.CREST.ScaledRotationalConstantSettings.RotationalConstantThreshold = (
0.01 # default is 0.003
)
filter_job = ConformersJob(settings=s)
filter_job.run()
conformerset = filter_job.results.conformerset
print("Number of conformers: ", len(conformerset))
[09.04|12:26:52] JOB conformers STARTED
[09.04|12:26:52] Renaming job conformers to conformers.003
[09.04|12:26:52] JOB conformers.003 RUNNING
[09.04|12:27:05] JOB conformers.003 FINISHED
[09.04|12:27:05] JOB conformers.003 SUCCESSFUL
Number of conformers: 1555
Alternatively, a filtering method can be applied that uses more local comparisons. The native AMS duplicate filter uses the interatomic distance matrix and torsion angles to determine if two structures are duplicates. This approach may be more appropriate for systems with multiple molecules, because the method only takes into account intra-molecular changes. On the other hand, this type of filtering is more time consuming, while in most cases the effect on the conformer set will not be extreme.
s = plams.Settings()
s.input.ams.Task = "Filter"
s.input.ams.InputConformersSet = rkf
s.input.ams.Equivalence.Method = "AMS"
filter_job = ConformersJob(settings=s)
filter_job.run()
conformerset = filter_job.results.conformerset
print("Number of AMS conformers: ", len(conformerset))
[09.04|12:27:05] JOB conformers STARTED
[09.04|12:27:05] Renaming job conformers to conformers.004
[09.04|12:27:05] JOB conformers.004 RUNNING
[09.04|12:27:36] JOB conformers.004 FINISHED
[09.04|12:27:36] JOB conformers.004 SUCCESSFUL
Number of AMS conformers: 1674
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Conformer generation for multiple molecules
#
# Lets see how two alanine molecules orient themselves using CREST conformer generation.
# To do this we will constrain the system in a spherical region using the `SphericalWall` constraint.
# We start by setting up a system of two alanine molecules in a relatively small space.
# ## Initial imports
import sys
import random
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import scm.plams as plams
from scm.base import ChemicalSystem
from scm.conformers import ConformersJob
from scm.utils.conversions import chemsys_to_plams_molecule, plams_molecule_to_chemsys
# ## Alanine dimer
from scm.plams import preoptimize
smiles = "CC(N)C(=O)O.CC(N)C(=O)O"
dimer = ChemicalSystem.from_smiles(smiles) # alternatively you may use packmol to build the dimer
dimer.translate(-dimer.center_of_mass())
# Optimize the dimer structure prior to the conformer search.
s = plams.Settings()
s.input.GFNFF = plams.Settings()
s.input.ams.Task = "GeometryOptimization"
s.input.ams.GeometryOptimization.Convergence.Quality = "VeryGood"
s.input.ams.GeometryOptimization.Maxiterations = 1300
s.input.ams.GeometryOptimization.Method = "Quasi-Newton"
job = plams.AMSJob(molecule=dimer, settings=s)
job.run()
dimer = job.results.get_main_system()
plams.view(dimer, height=300, width=300, direction="along_pca3", picture_path="picture1.png")
# ## Calculation setup
# To determine the radius of the `SphericalWall` we measure the size of the initial dimer.
dists = dimer.distance_matrix()
max_dist = np.max(dists)
diameter = 1.33 * max_dist
radius = diameter / 2
print(f"Largest distance between atoms: {max_dist:.3f} ang.")
print(f"Radius: {radius:.3f} ang.")
# Now we can set up the Crest conformer generation job, with the appropriate spherical wall constraining the molecules close together.
# This seed will used for the starting MD velocities in conformers generation
# Due to numerical aspects it does not guarantee full reproducibility.
seed = random.randint(1, 10000000)
print(f"Seed used for stochastic aspects is {seed}.")
settings = plams.Settings()
settings.input.ams.EngineAddons.WallPotential.Enabled = "Yes"
settings.input.ams.EngineAddons.WallPotential.Radius = radius
settings.input.ams.Generator.Method = "CREST"
settings.input.ams.Output.KeepWorkDir = "Yes"
settings.input.ams.GeometryOptimization.MaxConvergenceTime = "High"
settings.input.ams.Generator.CREST.NCycles = 3 # at most 3 CREST cycles for this demo
settings.input.ams.Generator.RNGSeed = seed
settings.input.ams.Generator.CREST.NMolDynStepsFactor = 3
settings.input.GFNFF = plams.Settings()
# ## Run the conformers job
# Now we can run the conformer generation job. This job will run somewhere between 30 minutes and 1 hour.
job = ConformersJob(molecule=chemsys_to_plams_molecule(dimer), settings=settings)
job.run()
# ConformersJob.load_external("plams_workdir/conformers/conformers.rkf") # load from disk instead of running the job
# Now, remove the wall and reoptimize
if "EngineAddons" in settings.input.ams:
del settings.input.ams.EngineAddons
settings.input.ams.Task = "Optimize"
settings.input.ams.InputConformersSet = job.results.rkfpath()
opt_job = ConformersJob(settings=settings)
opt_job.run()
rkf = opt_job.results.rkfpath()
print(f"Conformers stored in {rkf}")
# ## Results
relative_energies = opt_job.results.get_relative_energies("kcal/mol")
print(f"Total number of conformers: {len(relative_energies)}")
conformer_table = pd.DataFrame(
{
"Conformer Id": np.arange(1, len(relative_energies) + 1),
"Rel. E (kcal/mol)": relative_energies,
"Population at 298 K": opt_job.results.get_boltzmann_distribution(298),
}
)
print(conformer_table.head(20))
# Here we plot the three lowest-energy conformers.
from scm.conformers.plams.plot import plot_conformers
axes = plot_conformers(opt_job, width=300, height=300, direction="along_pca3")
axes
import matplotlib.pyplot as plt
plt.gcf().savefig("picture2.png")
# You can also open the conformers in AMSmovie to browse all conformers 500+ conformers:
# get_ipython().system('amsmovie "{opt_job.results.rkfpath()}"')
# ### Filtering out duplicates
# The `conformerset` attribute holds information about the full set of conformers.
conformerset = opt_job.results.conformerset
print("Number of conforrmers: ", len(conformerset))
# This conformerset does not contain any duplicates. However, there is no single definition of the term duplicates when applied to conformers. By default, duplicates are defined based on energy and rotational constants, the latter quantifying the 3D shape of the system. The filtering method uses thresholds for energy and rotational constants, which have been thoroughly tested for (mostly small) single molecules systems. If the user feels that the stored conformers are too similar, these thresholds can of course be changed. Below we refilter the stored conformerset using more lenient thresholds (systems with greater differences will be considered duplicates).
s = plams.Settings()
s.input.ams.Task = "Filter"
s.input.ams.InputConformersSet = rkf
s.input.ams.Equivalence.CREST.EnergyThreshold = 0.2 # default is 0.05
s.input.ams.Equivalence.CREST.ScaledRotationalConstantSettings.RotationalConstantThreshold = 0.01 # default is 0.003
filter_job = ConformersJob(settings=s)
filter_job.run()
conformerset = filter_job.results.conformerset
print("Number of conformers: ", len(conformerset))
# Alternatively, a filtering method can be applied that uses more local comparisons. The native AMS duplicate filter uses the interatomic distance matrix and torsion angles to determine if two structures are duplicates. This approach may be more appropriate for systems with multiple molecules, because the method only takes into account intra-molecular changes. On the other hand, this type of filtering is more time consuming, while in most cases the effect on the conformer set will not be extreme.
s = plams.Settings()
s.input.ams.Task = "Filter"
s.input.ams.InputConformersSet = rkf
s.input.ams.Equivalence.Method = "AMS"
filter_job = ConformersJob(settings=s)
filter_job.run()
conformerset = filter_job.results.conformerset
print("Number of AMS conformers: ", len(conformerset))