Tuning the Range Separation Parameter in ADF¶
In this example we optimize the value of gamma parameter for long-range corrected XC functional (in our case: LCY-PBE) in ADF. Long-range corrected XC functionals can be used in ADF with XCfun.
The optimal range separation parameter gamma yields the HOMO energy equal to the ionization potential (IP). Given a molecular system, we simultaneously minimize the difference between HOMO and IP for that system (N) and its anion (A) (system with one more electron). We define the J function as:
and find the value of gamma (within a certain range) which minimizes J. See also this article by Kronik and coworkers.
We first define a new job type GammaJob by extending MultiJob.
The goal of GammaJob is to calculate the J function for one fixed value of gamma
To do that we need to perform 3 different single point calculations: 1 for the given system (let’s call it S0), 1 for the system with one more electron (S-) and 1 for the system with one less electron (S+).
S+ calculation is needed to find the ionization potential of S0.
The constructor (__init__) of GammaJob accepts several new arguments and simply stores them.
These new arguments define: the value of gamma, the Molecule together with its initial charge, and the values of spin for S-, S0 and S+ (as a tuple of length 3).
Then the prerun method is used to create three children jobs with different values of total charge and spin multiplicity.
A dedicated Results subclass features a simple method for extracting the value of J based on results on three children jobs.
We can then treat our newly defined GammaJob as a blackbox with simple interface: input gamma -> run -> extract J.
The next step is to create multiple instances of GammaJob for a range of different gammas.
That task can be conveniently wrapped in a simple function gamma_scan.
Tuning the range separation¶
This example tunes the range-separation parameter gamma for a long-range corrected functional in ADF. For each gamma value, it evaluates the J function that combines HOMO-IP differences for neutral and anionic states, then selects the gamma that minimizes J.
Initial Imports¶
import psutil
from typing import List, Sequence, Tuple
import matplotlib.pyplot as plt
import numpy as np
from matplotlib.axes import Axes
from scm.plams import (
AMSJob,
Atom,
JobRunner,
Molecule,
MultiJob,
Results,
Settings,
config,
view,
)
Define Helper Job and Result Classes¶
A GammaJob runs three single-point calculations for one gamma value (charge -1, 0, +1), and GammaResults computes the corresponding J value.
class GammaResults(Results):
@staticmethod
def get_difference(job: AMSJob, job_plus: AMSJob) -> float:
"""Return HOMO + (E_plus - E) for one charge state pair."""
homo = float(job.results.readrkf("Properties", "HOMO", file="engine"))
ionization = float(job_plus.results.get_energy() - job.results.get_energy())
return ionization + homo
def get_J(self) -> float:
n_term = GammaResults.get_difference(self.job.children[1], self.job.children[2])
a_term = GammaResults.get_difference(self.job.children[0], self.job.children[1])
return float((n_term**2 + a_term**2) ** 0.5)
class GammaJob(MultiJob):
_result_type = GammaResults
def __init__(
self,
molecule: Molecule,
gamma: float,
charge: int,
spins: Tuple[int, int, int],
**kwargs,
) -> None:
super().__init__(**kwargs)
self.molecule = molecule
self.gamma = gamma
self.charge = charge
self.spins = spins
def prerun(self) -> None:
charges = [self.charge - 1, self.charge, self.charge + 1]
for charge, spin in zip(charges, self.spins):
name = f"{self.name}_charge_{charge}".replace("-", "minus")
child = AMSJob(name=name, molecule=self.molecule, settings=self.settings)
child.molecule.properties.charge = charge
child.settings.input.adf.xc.rangesep = f"gamma={self.gamma:f}"
if spin != 0:
child.settings.input.adf.unrestricted = True
child.settings.input.adf.SpinPolarization = spin
self.children.append(child)
def gamma_scan(
gammas: Sequence[float],
settings: Settings,
molecule: Molecule,
name: str = "scan",
charge: int = 0,
spins: Tuple[int, int, int] = (1, 0, 1),
) -> List[Tuple[float, float]]:
jobs = [
GammaJob(
molecule=molecule,
settings=settings,
gamma=float(gamma),
charge=charge,
spins=spins,
name=f"{name}_gamma_{gamma}",
)
for gamma in gammas
]
results = [job.run() for job in jobs]
j_values = [result.get_J() for result in results]
return list(zip([float(g) for g in gammas], j_values))
def plot_scan(scan_results: Sequence[Tuple[float, float]]) -> Axes:
fig, ax = plt.subplots(figsize=(6, 4))
gammas = [gamma for gamma, _ in scan_results]
j_values = [j for _, j in scan_results]
ax.plot(gammas, j_values, marker="o")
ax.set_xlabel("gamma")
ax.set_ylabel("J")
fig.tight_layout()
return ax
Run Gamma Scan¶
Configure PLAMS to run independent gamma points in parallel.
config.default_jobrunner = JobRunner(parallel=True, maxjobs=psutil.cpu_count(logical=False))
Set up ADF single-point settings with PBE and XCfun enabled.
s = Settings()
s.input.ams.task = "SinglePoint"
s.input.adf.basis.type = "DZP"
s.input.adf.basis.core = "None"
s.input.adf.xc.gga = "PBE"
s.input.adf.xc.xcfun = True
s.runscript.nproc = 1
Build a toy H2 molecule for the scan.
mol = Molecule()
mol.add_atom(Atom(symbol="H", coords=(0.0, 0.0, -0.3540)))
mol.add_atom(Atom(symbol="H", coords=(0.0, 0.0, 0.3540)))
view(mol, width=250, height=250, direction="along_x")
[10.04|13:56:32] Starting Xvfb...
[10.04|13:56:32] Xvfb started

Run a small gamma scan. For production use, use a wider range and finer spacing.
gammas = np.around(np.arange(1.2, 1.9, 0.1), decimals=3)
scan_results = gamma_scan(gammas, s, mol, name="gamma_scan")
[10.04|13:56:33] JOB gamma_scan_gamma_1.2 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.3 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.4 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.5 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.6 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.7 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.2 RUNNING
[10.04|13:56:33] JOB gamma_scan_gamma_1.2/gamma_scan_gamma_1.2_charge_minus1 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.8 STARTED
[10.04|13:56:33] JOB gamma_scan_gamma_1.2/gamma_scan_gamma_1.2_charge_0 STARTED
... output trimmed ....
[10.04|13:56:53] JOB gamma_scan_gamma_1.7 FINISHED
[10.04|13:56:53] JOB gamma_scan_gamma_1.6 FINISHED
[10.04|13:56:53] Pickling of gamma_scan_gamma_1.3 failed
[10.04|13:56:53] JOB gamma_scan_gamma_1.3 SUCCESSFUL
[10.04|13:56:53] Pickling of gamma_scan_gamma_1.6 failed
[10.04|13:56:53] Pickling of gamma_scan_gamma_1.8 failed
[10.04|13:56:53] JOB gamma_scan_gamma_1.6 SUCCESSFUL
[10.04|13:56:53] JOB gamma_scan_gamma_1.8 SUCCESSFUL
[10.04|13:56:53] Pickling of gamma_scan_gamma_1.7 failed
[10.04|13:56:53] JOB gamma_scan_gamma_1.7 SUCCESSFUL
###
print("== Results ==")
print("gamma J")
for gamma, j_value in scan_results:
print(f"{gamma:.4f} {j_value:.8f}")
best_gamma = min(scan_results, key=lambda pair: pair[1])[0]
print(f"Optimal gamma value: {best_gamma:.4f}")
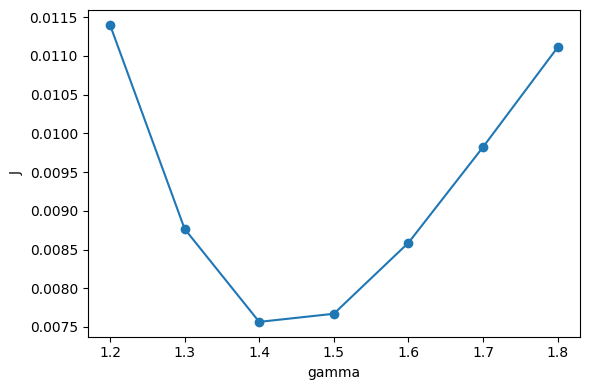
== Results ==
gamma J
1.2000 0.01139992
1.3000 0.00876582
1.4000 0.00756612
1.5000 0.00767076
1.6000 0.00858674
1.7000 0.00982228
1.8000 0.01110911
Optimal gamma value: 1.4000
Plot J as a function of gamma and identify the minimum.
ax = plot_scan(scan_results)
ax;
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Tuning the range separation
#
# This example tunes the range-separation parameter `gamma` for a long-range corrected functional in ADF. For each gamma value, it evaluates the J function that combines HOMO-IP differences for neutral and anionic states, then selects the gamma that minimizes J.
#
# ### Initial Imports
import psutil
from typing import List, Sequence, Tuple
import matplotlib.pyplot as plt
import numpy as np
from matplotlib.axes import Axes
from scm.plams import (
AMSJob,
Atom,
JobRunner,
Molecule,
MultiJob,
Results,
Settings,
config,
view,
)
# ### Define Helper Job and Result Classes
#
# A `GammaJob` runs three single-point calculations for one gamma value (charge -1, 0, +1), and `GammaResults` computes the corresponding J value.
class GammaResults(Results):
@staticmethod
def get_difference(job: AMSJob, job_plus: AMSJob) -> float:
"""Return HOMO + (E_plus - E) for one charge state pair."""
homo = float(job.results.readrkf("Properties", "HOMO", file="engine"))
ionization = float(job_plus.results.get_energy() - job.results.get_energy())
return ionization + homo
def get_J(self) -> float:
n_term = GammaResults.get_difference(self.job.children[1], self.job.children[2])
a_term = GammaResults.get_difference(self.job.children[0], self.job.children[1])
return float((n_term**2 + a_term**2) ** 0.5)
class GammaJob(MultiJob):
_result_type = GammaResults
def __init__(
self,
molecule: Molecule,
gamma: float,
charge: int,
spins: Tuple[int, int, int],
**kwargs,
) -> None:
super().__init__(**kwargs)
self.molecule = molecule
self.gamma = gamma
self.charge = charge
self.spins = spins
def prerun(self) -> None:
charges = [self.charge - 1, self.charge, self.charge + 1]
for charge, spin in zip(charges, self.spins):
name = f"{self.name}_charge_{charge}".replace("-", "minus")
child = AMSJob(name=name, molecule=self.molecule, settings=self.settings)
child.molecule.properties.charge = charge
child.settings.input.adf.xc.rangesep = f"gamma={self.gamma:f}"
if spin != 0:
child.settings.input.adf.unrestricted = True
child.settings.input.adf.SpinPolarization = spin
self.children.append(child)
def gamma_scan(
gammas: Sequence[float],
settings: Settings,
molecule: Molecule,
name: str = "scan",
charge: int = 0,
spins: Tuple[int, int, int] = (1, 0, 1),
) -> List[Tuple[float, float]]:
jobs = [
GammaJob(
molecule=molecule,
settings=settings,
gamma=float(gamma),
charge=charge,
spins=spins,
name=f"{name}_gamma_{gamma}",
)
for gamma in gammas
]
results = [job.run() for job in jobs]
j_values = [result.get_J() for result in results]
return list(zip([float(g) for g in gammas], j_values))
def plot_scan(scan_results: Sequence[Tuple[float, float]]) -> Axes:
fig, ax = plt.subplots(figsize=(6, 4))
gammas = [gamma for gamma, _ in scan_results]
j_values = [j for _, j in scan_results]
ax.plot(gammas, j_values, marker="o")
ax.set_xlabel("gamma")
ax.set_ylabel("J")
fig.tight_layout()
return ax
# ### Run Gamma Scan
#
# Configure PLAMS to run independent gamma points in parallel.
config.default_jobrunner = JobRunner(parallel=True, maxjobs=psutil.cpu_count(logical=False))
# Set up ADF single-point settings with PBE and XCfun enabled.
#
s = Settings()
s.input.ams.task = "SinglePoint"
s.input.adf.basis.type = "DZP"
s.input.adf.basis.core = "None"
s.input.adf.xc.gga = "PBE"
s.input.adf.xc.xcfun = True
s.runscript.nproc = 1
# Build a toy H2 molecule for the scan.
#
mol = Molecule()
mol.add_atom(Atom(symbol="H", coords=(0.0, 0.0, -0.3540)))
mol.add_atom(Atom(symbol="H", coords=(0.0, 0.0, 0.3540)))
view(mol, width=250, height=250, direction="along_x", picture_path="picture1.png")
# Run a small gamma scan. For production use, use a wider range and finer spacing.
#
gammas = np.around(np.arange(1.2, 1.9, 0.1), decimals=3)
scan_results = gamma_scan(gammas, s, mol, name="gamma_scan")
###
print("== Results ==")
print("gamma J")
for gamma, j_value in scan_results:
print(f"{gamma:.4f} {j_value:.8f}")
best_gamma = min(scan_results, key=lambda pair: pair[1])[0]
print(f"Optimal gamma value: {best_gamma:.4f}")
# Plot J as a function of gamma and identify the minimum.
#
ax = plot_scan(scan_results)
ax
ax.figure.savefig("picture2.png")