BSSE correction for H2O on ZnO(10-10)¶
Calculate basis set superposition error (BSSE) with BAND using the counterpoise correction workflow for water adsorption on ZnO(10-10).
Run counterpoise calculations for multiple basis sets
Compare uncorrected and counterpoise-corrected adsorption energies
Plot the adsorption-energy trends versus basis quality
BSSE correction for H2O adsorption on ZnO(10-10)¶
This example reproduces the BSSE analysis from the Crystals and Surfaces tutorial. It runs BAND counterpoise calculations for five basis sets and plots corrected vs. uncorrected adsorption energies.
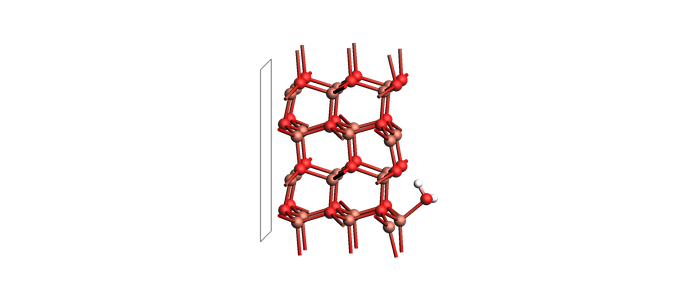
Define the slab+molecule system¶
The full structure is embedded below as an AMS System block, so this notebook does not depend on external files. Atoms 1, 2, and 3 are the adsorbate H2O fragment used in the counterpoise setup.
from typing import Dict, List, Tuple
import matplotlib.pyplot as plt
import scm.plams as plams
from scm.base import ChemicalSystem
from scm.plams import Settings, view
from scm.plams.recipes.counterpoise import CounterpoiseEnergyJob
from scm.utils.conversions import chemsys_to_plams_molecule
ab_system = ChemicalSystem(
"""
System
Atoms
H 4.194124 2.321011 9.990591
O 3.277064 2.369351 9.662700
H 3.215647 3.302746 9.160403
O 3.121052 4.371977 8.152437
O 3.157940 9.599596 8.160532
O -0.155188 4.377678 8.158283
O -0.092407 9.610401 8.171480
Zn 3.135686 1.046635 8.068212
Zn 3.127411 6.251538 7.820969
Zn -0.123253 6.228628 7.789648
Zn -0.108077 0.986306 7.727521
Zn 1.540445 9.053723 7.248234
Zn 4.781682 9.052463 7.248636
Zn 1.454711 3.779398 7.208485
Zn 4.770754 3.793547 7.144487
O 1.537161 1.798390 7.074007
O 4.752079 1.794734 7.054472
O 1.506053 7.034856 7.081453
O 4.747135 7.034630 7.078047
O 1.504136 4.449837 5.256964
O 1.510233 9.664240 5.251216
O 4.745874 4.467809 5.219866
O 4.765413 9.642604 5.251871
Zn 1.527610 1.213182 5.119746
Zn 4.738978 1.206559 5.113718
Zn 1.504703 6.460518 5.092362
Zn 4.750778 6.477069 5.090083
Zn -0.110739 9.072159 4.273393
Zn 3.135224 9.070253 4.279334
Zn -0.117138 3.823484 4.274300
Zn 3.118895 3.832597 4.260451
O -0.115211 1.822988 4.141734
O 3.131372 1.834356 4.108967
O -0.117871 7.064012 4.120018
O 3.122850 7.064255 4.112580
O -0.120166 4.440964 2.316795
O -0.116391 9.670504 2.304567
O 3.130312 4.443558 2.298592
O 3.130933 9.674750 2.306875
Zn -0.116763 1.207088 2.147541
Zn 3.130704 1.213453 2.119764
Zn -0.119363 6.435899 2.118992
Zn 3.130940 6.437444 2.109042
Zn 4.758737 3.618851 1.589370
Zn 4.755815 8.858895 1.584735
Zn 1.508461 8.855081 1.581480
Zn 1.502167 3.625979 1.585735
O 4.763512 1.779351 1.204246
O 1.500332 1.785698 1.205474
O 4.757821 7.015026 1.196018
O 1.503517 7.011011 1.191741
End
Lattice
6.50000095 0.0 0.0
0.0 10.46000194 0.0
End
End
"""
)
view(ab_system, width=700, height=300, direction="tilt_x", guess_bonds=True)
Run counterpoise jobs for each basis set¶
This section can take significant time. For production use, run on appropriate compute resources.
ab_mol = chemsys_to_plams_molecule(ab_system)
ids_state_A: List[int] = [1, 2, 3] # H2O fragment
basis_sets: List[str] = ["DZ", "DZP", "TZP", "TZ2P", "QZ4P"]
base_settings = Settings()
base_settings.input.ams.Task = "GeometryOptimization"
base_settings.input.band.KSpace.Quality = "Basic"
base_settings.input.band.XC.GGA = "PBE"
jobs: Dict[str, CounterpoiseEnergyJob] = {}
for basis in basis_sets:
settings = base_settings.copy()
settings.input.band.Basis.Type = basis
job = CounterpoiseEnergyJob(
name=f"{basis}_counterpoise",
AB=ab_mol.copy(),
ids_state_A=ids_state_A,
settings_state_AB=settings,
)
job.run()
jobs[basis] = job
[13.03|12:35:35] JOB DZ_counterpoise STARTED
[13.03|12:35:35] JOB DZ_counterpoise RUNNING
[13.03|12:35:35] JOB DZ_counterpoise/AB_geo_AB_basis_AB STARTED
[13.03|12:35:35] JOB DZ_counterpoise/AB_geo_AB_basis_AB RUNNING
[13.03|12:56:26] JOB DZ_counterpoise/AB_geo_AB_basis_AB FINISHED
[13.03|12:56:26] JOB DZ_counterpoise/AB_geo_AB_basis_AB SUCCESSFUL
[13.03|12:56:26] JOB DZ_counterpoise/A_geo_AB_basis_AB STARTED
[13.03|12:56:26] JOB DZ_counterpoise/A_geo_AB_basis_AB RUNNING
[13.03|12:57:05] JOB DZ_counterpoise/A_geo_AB_basis_AB FINISHED
[13.03|12:57:05] JOB DZ_counterpoise/A_geo_AB_basis_AB SUCCESSFUL
[13.03|12:57:05] JOB DZ_counterpoise/A_geo_AB_basis_A STARTED
[13.03|12:57:05] JOB DZ_counterpoise/A_geo_AB_basis_A RUNNING
... output trimmed ....
[13.03|18:38:03] JOB QZ4P_counterpoise/B_geo_AB_basis_B FINISHED
[13.03|18:38:03] JOB QZ4P_counterpoise/B_geo_AB_basis_B SUCCESSFUL
[13.03|18:38:03] JOB QZ4P_counterpoise/A_geo_A_basis_A STARTED
[13.03|18:38:03] JOB QZ4P_counterpoise/A_geo_A_basis_A RUNNING
[13.03|18:40:15] JOB QZ4P_counterpoise/A_geo_A_basis_A FINISHED
[13.03|18:40:15] JOB QZ4P_counterpoise/A_geo_A_basis_A SUCCESSFUL
[13.03|18:40:15] JOB QZ4P_counterpoise/B_geo_B_basis_B STARTED
[13.03|18:40:15] JOB QZ4P_counterpoise/B_geo_B_basis_B RUNNING
[13.03|20:23:50] JOB QZ4P_counterpoise/B_geo_B_basis_B FINISHED
[13.03|20:23:50] JOB QZ4P_counterpoise/B_geo_B_basis_B SUCCESSFUL
[13.03|20:23:50] JOB QZ4P_counterpoise FINISHED
[13.03|20:23:50] JOB QZ4P_counterpoise SUCCESSFUL
rows: List[Tuple[str, float, float, float]] = []
for basis in basis_sets:
energies = jobs[basis].results.get_all_energies(unit="kcal/mol")
rows.append((basis, energies["Ebind_raw"], energies["Ebind_cp"], -energies["BSSE_tot"]))
print("Basis Ebind_uncorrected Ebind_corrected Counterpoise_correction")
for basis, raw, corrected, cp_corr in rows:
print(f"{basis:5s} {raw:17.2f} {corrected:15.2f} {cp_corr:24.2f}")
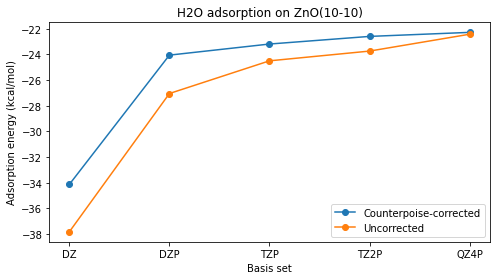
Basis Ebind_uncorrected Ebind_corrected Counterpoise_correction
DZ -37.83 -34.13 3.70
DZP -27.05 -24.05 2.99
TZP -24.50 -23.19 1.31
TZ2P -23.74 -22.59 1.14
QZ4P -22.43 -22.28 0.14
Plot adsorption energies¶
labels = [r[0] for r in rows]
uncorrected = [r[1] for r in rows]
corrected = [r[2] for r in rows]
x = list(range(len(labels)))
fig, ax = plt.subplots(figsize=(7, 4))
ax.plot(x, corrected, "o-", label="Counterpoise-corrected")
ax.plot(x, uncorrected, "o-", label="Uncorrected")
ax.set_title("H2O adsorption on ZnO(10-10)")
ax.set_xlabel("Basis set")
ax.set_ylabel("Adsorption energy (kcal/mol)")
ax.set_xticks(x)
ax.set_xticklabels(labels)
ax.legend()
fig.tight_layout()
ax;
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## BSSE correction for H2O adsorption on ZnO(10-10)
#
# This example reproduces the BSSE analysis from the *Crystals and Surfaces* tutorial.
# It runs BAND counterpoise calculations for five basis sets and plots corrected vs. uncorrected adsorption energies.
#
# ## Define the slab+molecule system
#
# The full structure is embedded below as an AMS `System` block, so this notebook does not depend on external files.
# Atoms 1, 2, and 3 are the adsorbate H2O fragment used in the counterpoise setup.
#
from typing import Dict, List, Tuple
import matplotlib.pyplot as plt
import scm.plams as plams
from scm.base import ChemicalSystem
from scm.plams import Settings, view
from scm.plams.recipes.counterpoise import CounterpoiseEnergyJob
from scm.utils.conversions import chemsys_to_plams_molecule
ab_system = ChemicalSystem(
"""
System
Atoms
H 4.194124 2.321011 9.990591
O 3.277064 2.369351 9.662700
H 3.215647 3.302746 9.160403
O 3.121052 4.371977 8.152437
O 3.157940 9.599596 8.160532
O -0.155188 4.377678 8.158283
O -0.092407 9.610401 8.171480
Zn 3.135686 1.046635 8.068212
Zn 3.127411 6.251538 7.820969
Zn -0.123253 6.228628 7.789648
Zn -0.108077 0.986306 7.727521
Zn 1.540445 9.053723 7.248234
Zn 4.781682 9.052463 7.248636
Zn 1.454711 3.779398 7.208485
Zn 4.770754 3.793547 7.144487
O 1.537161 1.798390 7.074007
O 4.752079 1.794734 7.054472
O 1.506053 7.034856 7.081453
O 4.747135 7.034630 7.078047
O 1.504136 4.449837 5.256964
O 1.510233 9.664240 5.251216
O 4.745874 4.467809 5.219866
O 4.765413 9.642604 5.251871
Zn 1.527610 1.213182 5.119746
Zn 4.738978 1.206559 5.113718
Zn 1.504703 6.460518 5.092362
Zn 4.750778 6.477069 5.090083
Zn -0.110739 9.072159 4.273393
Zn 3.135224 9.070253 4.279334
Zn -0.117138 3.823484 4.274300
Zn 3.118895 3.832597 4.260451
O -0.115211 1.822988 4.141734
O 3.131372 1.834356 4.108967
O -0.117871 7.064012 4.120018
O 3.122850 7.064255 4.112580
O -0.120166 4.440964 2.316795
O -0.116391 9.670504 2.304567
O 3.130312 4.443558 2.298592
O 3.130933 9.674750 2.306875
Zn -0.116763 1.207088 2.147541
Zn 3.130704 1.213453 2.119764
Zn -0.119363 6.435899 2.118992
Zn 3.130940 6.437444 2.109042
Zn 4.758737 3.618851 1.589370
Zn 4.755815 8.858895 1.584735
Zn 1.508461 8.855081 1.581480
Zn 1.502167 3.625979 1.585735
O 4.763512 1.779351 1.204246
O 1.500332 1.785698 1.205474
O 4.757821 7.015026 1.196018
O 1.503517 7.011011 1.191741
End
Lattice
6.50000095 0.0 0.0
0.0 10.46000194 0.0
End
End
"""
)
view(ab_system, width=700, height=300, direction="tilt_x", guess_bonds=True, picture_path="picture1.png")
# ## Run counterpoise jobs for each basis set
#
# This section can take significant time. For production use, run on appropriate compute resources.
#
ab_mol = chemsys_to_plams_molecule(ab_system)
ids_state_A: List[int] = [1, 2, 3] # H2O fragment
basis_sets: List[str] = ["DZ", "DZP", "TZP", "TZ2P", "QZ4P"]
base_settings = Settings()
base_settings.input.ams.Task = "GeometryOptimization"
base_settings.input.band.KSpace.Quality = "Basic"
base_settings.input.band.XC.GGA = "PBE"
jobs: Dict[str, CounterpoiseEnergyJob] = {}
for basis in basis_sets:
settings = base_settings.copy()
settings.input.band.Basis.Type = basis
job = CounterpoiseEnergyJob(
name=f"{basis}_counterpoise",
AB=ab_mol.copy(),
ids_state_A=ids_state_A,
settings_state_AB=settings,
)
job.run()
jobs[basis] = job
rows: List[Tuple[str, float, float, float]] = []
for basis in basis_sets:
energies = jobs[basis].results.get_all_energies(unit="kcal/mol")
rows.append((basis, energies["Ebind_raw"], energies["Ebind_cp"], -energies["BSSE_tot"]))
print("Basis Ebind_uncorrected Ebind_corrected Counterpoise_correction")
for basis, raw, corrected, cp_corr in rows:
print(f"{basis:5s} {raw:17.2f} {corrected:15.2f} {cp_corr:24.2f}")
# ## Plot adsorption energies
#
labels = [r[0] for r in rows]
uncorrected = [r[1] for r in rows]
corrected = [r[2] for r in rows]
x = list(range(len(labels)))
fig, ax = plt.subplots(figsize=(7, 4))
ax.plot(x, corrected, "o-", label="Counterpoise-corrected")
ax.plot(x, uncorrected, "o-", label="Uncorrected")
ax.set_title("H2O adsorption on ZnO(10-10)")
ax.set_xlabel("Basis set")
ax.set_ylabel("Adsorption energy (kcal/mol)")
ax.set_xticks(x)
ax.set_xticklabels(labels)
ax.legend()
fig.tight_layout()
ax
ax.figure.savefig("picture2.png")