Substituting Functional Groups in Molecules¶
Build functionalized molecules by adding ligands to a substrate, then generate and visualize the resulting products in 3D and 2D.
Initial imports¶
import scm.plams as plams
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import AllChem
from typing import List, Tuple
IPythonConsole.ipython_useSVG = True
IPythonConsole.molSize = 250, 250
Helper class and function¶
The MoleculeConnector class and substitute() method below are convenient to use.
class MoleculeConnector:
def __init__(self, molecule, connector, name="molecule"):
self.name = name
self.molecule = molecule
self.molecule.properties.name = name
self.connector = connector # 2-tuple of integers, unlike the Molecule.substitute() method which uses two atoms
def __str__(self):
return f"""
Name: {self.name}
{self.molecule}
Connector: {self.connector}. This means that when substitution is performed atom {self.connector[0]} will be kept in the substituted molecule. Atom {self.connector[1]}, and anything connected to it, will NOT be kept.
"""
def substitute(substrate: MoleculeConnector, ligand: MoleculeConnector):
"""
Returns: Molecule with the ligand added to the substrate, replacing the respective connector bonds.
"""
molecule = substrate.molecule.copy()
molecule.substitute(
connector=(molecule[substrate.connector[0]], molecule[substrate.connector[1]]),
ligand=ligand.molecule,
ligand_connector=(
ligand.molecule[ligand.connector[0]],
ligand.molecule[ligand.connector[1]],
),
)
return molecule
def set_atom_indices(rdmol: Chem.rdchem.Mol, start=0):
for atom in rdmol.GetAtoms():
atom.SetAtomMapNum(atom.GetIdx() + start) # give 1-based index
return rdmol
def to_lewis(molecule: plams.Molecule, template=None, regenerate: bool = True):
if isinstance(molecule, Chem.rdchem.Mol):
rdmol = molecule
else:
rdmol = plams.to_rdmol(molecule)
if regenerate:
rdmol = Chem.RemoveHs(rdmol)
smiles = Chem.MolToSmiles(rdmol)
rdmol = Chem.MolFromSmiles(smiles)
if template is not None:
AllChem.GenerateDepictionMatching2DStructure(rdmol, template)
try:
if molecule.properties.name:
rdmol.SetProp("_Name", molecule.properties.name)
except AttributeError:
pass
return rdmol
def smiles2template(smiles: str):
template = Chem.MolFromSmiles(smiles)
AllChem.Compute2DCoords(template)
return template
def draw_lewis_grid(
molecules: List[plams.Molecule],
molsPerRow: int = 4,
template_smiles: str = None,
regenerate: bool = False,
draw_atom_indices: bool = False,
draw_legend: bool = True,
):
template = None
if template_smiles:
template = smiles2template(template_smiles)
rdmols = [to_lewis(x, template=template, regenerate=regenerate) for x in molecules]
if draw_atom_indices:
for rdmol in rdmols:
set_atom_indices(rdmol, start=1)
legends = None
if draw_legend:
try:
legends = [x.properties.name or f"mol{i}" for i, x in enumerate(molecules)]
except AttributeError:
pass
return Draw.MolsToGridImage(rdmols, molsPerRow=molsPerRow, legends=legends)
Generate substrate molecule¶
substrate_smiles = "c1ccccc1"
substrate = plams.from_smiles(substrate_smiles, forcefield="uff")
substrate.properties.name = "benzene"
Find out which bond to cleave¶
In the molecule you need to define which bond to cleave. To find out the bonds, run for example:
for b in substrate.bonds:
el1 = b.atom1.symbol
el2 = b.atom2.symbol
idx1, idx2 = substrate.index(b)
print(f"{el1}({idx1})--{el2}({idx2})")
C(1)--C(2)
C(2)--C(3)
C(3)--C(4)
C(4)--C(5)
C(5)--C(6)
C(6)--C(1)
C(1)--H(7)
C(2)--H(8)
C(3)--H(9)
C(4)--H(10)
C(5)--H(11)
C(6)--H(12)
to find that atoms 6 (C) and 12 (H) are bonded. We will choose this bond to cleave.
Alternatively, we can inspect the molecule inside a Jupyter notebook, visualizing with AMSView, to also find that atoms 6 (C) and 12 (H) are bonded.
plams.view(
substrate, padding=-0.5, show_atom_labels=True, atom_label_type="Name", width=300, height=300
)
[10.04|11:46:11] Starting Xvfb...
[10.04|11:46:11] Xvfb started
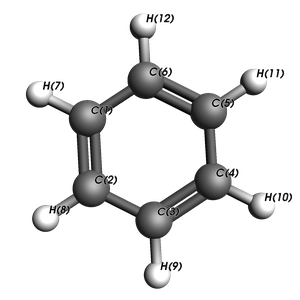
substrate_connector = MoleculeConnector(
substrate, (6, 12), "phenyl"
) # benzene becomes phenyl when bond between atoms 6,12 is cleaved
Define ligands¶
Perform the same steps for the ligands.
Note: The ligands below have an extra hydrogen or even more atoms compared to the name that they’re given.
ligands = [
MoleculeConnector(
plams.from_smiles("CCOC(=O)C", forcefield="uff"), (3, 2), "acetate"
), # ethyl acetate, bond from O to C cleaved
MoleculeConnector(
plams.from_smiles("O=NO", forcefield="uff"), (3, 4), "nitrite"
), # nitrous acid, bond from O to H cleaved
MoleculeConnector(
plams.from_smiles("Cl", forcefield="uff"), (1, 2), "chloride"
), # hydrogen chloride, bond from Cl to H cleaved
MoleculeConnector(
plams.from_smiles("c1ccccc1", forcefield="uff"), (6, 12), "phenyl"
), # benzene, bond to C to H cleaved
]
ligand_molecules = [ligand.molecule for ligand in ligands]
images = {
ligand.name: plams.view(
ligand.molecule, width=400, show_atom_labels=True, atom_label_type="Name"
)
for ligand in ligands
}
plams.plot_image_grid(images);
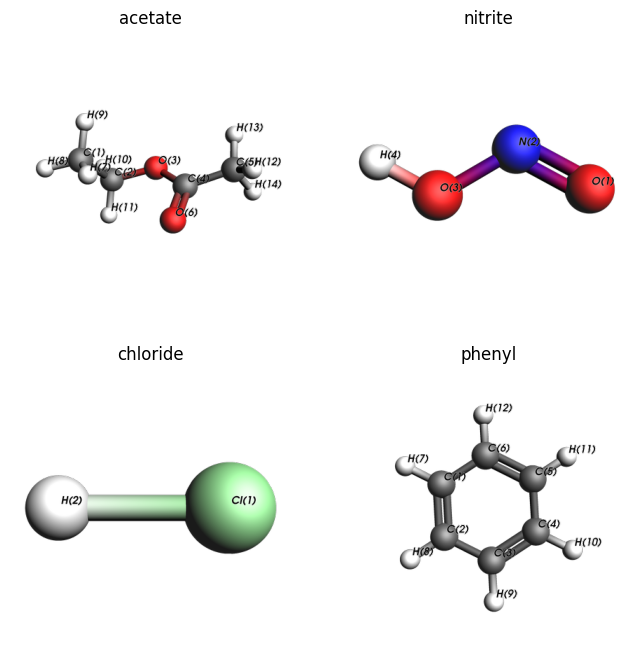
Above we see that cleaving the bonds from O(3)-C(2), O(3)-H(4), Cl(1)-H(2), and C(6)-H(12) will give the acetate, nitrite, chloride, and phenyl substituents, respectively.
Generate substituted molecules¶
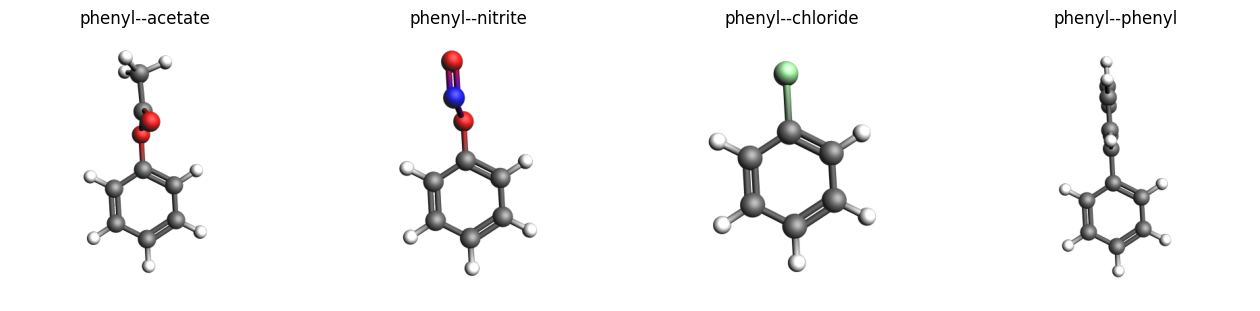
mols = []
for ligand in ligands:
mol = substitute(substrate_connector, ligand)
mol.properties.name = f"{substrate_connector.name}--{ligand.name}"
mols.append(mol)
print(f"Writing {mol.properties.name}.xyz")
mol.write(f"{mol.properties.name}.xyz")
print(f"{mol.properties.name} formula: {mol.get_formula(as_dict=True)}")
Writing phenyl--acetate.xyz
phenyl--acetate formula: {'C': 8, 'H': 8, 'O': 2}
Writing phenyl--nitrite.xyz
phenyl--nitrite formula: {'C': 6, 'H': 5, 'O': 2, 'N': 1}
Writing phenyl--chloride.xyz
phenyl--chloride formula: {'C': 6, 'H': 5, 'Cl': 1}
Writing phenyl--phenyl.xyz
phenyl--phenyl formula: {'C': 12, 'H': 10}
Plot 3D structures with PLAMS¶
images = {mol.properties.name: plams.view(mol, width=400) for mol in mols}
plams.plot_image_grid(images, rows=1);
Plot 2D Lewis structures with RDKit¶
The molecules can be aligned by using a benzene template. The regenerate option regenerates the molecule with RDkit to clean up the atomic positions.
draw_lewis_grid(mols, template_smiles=substrate_smiles, regenerate=True)

See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial imports
import scm.plams as plams
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import AllChem
from typing import List, Tuple
IPythonConsole.ipython_useSVG = True
IPythonConsole.molSize = 250, 250
# ## Helper class and function
#
# The ``MoleculeConnector`` class and ``substitute()`` method below are convenient to use.
class MoleculeConnector:
def __init__(self, molecule, connector, name="molecule"):
self.name = name
self.molecule = molecule
self.molecule.properties.name = name
self.connector = connector # 2-tuple of integers, unlike the Molecule.substitute() method which uses two atoms
def __str__(self):
return f"""
Name: {self.name}
{self.molecule}
Connector: {self.connector}. This means that when substitution is performed atom {self.connector[0]} will be kept in the substituted molecule. Atom {self.connector[1]}, and anything connected to it, will NOT be kept.
"""
def substitute(substrate: MoleculeConnector, ligand: MoleculeConnector):
"""
Returns: Molecule with the ligand added to the substrate, replacing the respective connector bonds.
"""
molecule = substrate.molecule.copy()
molecule.substitute(
connector=(molecule[substrate.connector[0]], molecule[substrate.connector[1]]),
ligand=ligand.molecule,
ligand_connector=(
ligand.molecule[ligand.connector[0]],
ligand.molecule[ligand.connector[1]],
),
)
return molecule
def set_atom_indices(rdmol: Chem.rdchem.Mol, start=0):
for atom in rdmol.GetAtoms():
atom.SetAtomMapNum(atom.GetIdx() + start) # give 1-based index
return rdmol
def to_lewis(molecule: plams.Molecule, template=None, regenerate: bool = True):
if isinstance(molecule, Chem.rdchem.Mol):
rdmol = molecule
else:
rdmol = plams.to_rdmol(molecule)
if regenerate:
rdmol = Chem.RemoveHs(rdmol)
smiles = Chem.MolToSmiles(rdmol)
rdmol = Chem.MolFromSmiles(smiles)
if template is not None:
AllChem.GenerateDepictionMatching2DStructure(rdmol, template)
try:
if molecule.properties.name:
rdmol.SetProp("_Name", molecule.properties.name)
except AttributeError:
pass
return rdmol
def smiles2template(smiles: str):
template = Chem.MolFromSmiles(smiles)
AllChem.Compute2DCoords(template)
return template
def draw_lewis_grid(
molecules: List[plams.Molecule],
molsPerRow: int = 4,
template_smiles: str = None,
regenerate: bool = False,
draw_atom_indices: bool = False,
draw_legend: bool = True,
):
template = None
if template_smiles:
template = smiles2template(template_smiles)
rdmols = [to_lewis(x, template=template, regenerate=regenerate) for x in molecules]
if draw_atom_indices:
for rdmol in rdmols:
set_atom_indices(rdmol, start=1)
legends = None
if draw_legend:
try:
legends = [x.properties.name or f"mol{i}" for i, x in enumerate(molecules)]
except AttributeError:
pass
return Draw.MolsToGridImage(rdmols, molsPerRow=molsPerRow, legends=legends)
# ## Generate substrate molecule
substrate_smiles = "c1ccccc1"
substrate = plams.from_smiles(substrate_smiles, forcefield="uff")
substrate.properties.name = "benzene"
# ## Find out which bond to cleave
# In the molecule you need to define which bond to cleave. To find out the bonds, run for example:
for b in substrate.bonds:
el1 = b.atom1.symbol
el2 = b.atom2.symbol
idx1, idx2 = substrate.index(b)
print(f"{el1}({idx1})--{el2}({idx2})")
# to find that atoms 6 (C) and 12 (H) are bonded. We will choose this bond to cleave.
#
# Alternatively, we can inspect the molecule inside a Jupyter notebook, visualizing with AMSView, to also find that atoms 6 (C) and 12 (H) are bonded.
plams.view(
substrate,
padding=-0.5,
show_atom_labels=True,
atom_label_type="Name",
width=300,
height=300,
picture_path="picture1.png",
)
substrate_connector = MoleculeConnector(
substrate, (6, 12), "phenyl"
) # benzene becomes phenyl when bond between atoms 6,12 is cleaved
# ## Define ligands
# Perform the same steps for the ligands.
#
# **Note**: The ligands below have an extra hydrogen or even more atoms compared to the name that they're given.
ligands = [
MoleculeConnector(
plams.from_smiles("CCOC(=O)C", forcefield="uff"), (3, 2), "acetate"
), # ethyl acetate, bond from O to C cleaved
MoleculeConnector(
plams.from_smiles("O=NO", forcefield="uff"), (3, 4), "nitrite"
), # nitrous acid, bond from O to H cleaved
MoleculeConnector(
plams.from_smiles("Cl", forcefield="uff"), (1, 2), "chloride"
), # hydrogen chloride, bond from Cl to H cleaved
MoleculeConnector(
plams.from_smiles("c1ccccc1", forcefield="uff"), (6, 12), "phenyl"
), # benzene, bond to C to H cleaved
]
ligand_molecules = [ligand.molecule for ligand in ligands]
images = {
ligand.name: plams.view(ligand.molecule, width=400, show_atom_labels=True, atom_label_type="Name")
for ligand in ligands
}
plams.plot_image_grid(images, save_path="picture2.png")
# Above we see that cleaving the bonds from O(3)-C(2), O(3)-H(4), Cl(1)-H(2), and C(6)-H(12) will give the acetate, nitrite, chloride, and phenyl substituents, respectively.
# ## Generate substituted molecules
mols = []
for ligand in ligands:
mol = substitute(substrate_connector, ligand)
mol.properties.name = f"{substrate_connector.name}--{ligand.name}"
mols.append(mol)
print(f"Writing {mol.properties.name}.xyz")
mol.write(f"{mol.properties.name}.xyz")
print(f"{mol.properties.name} formula: {mol.get_formula(as_dict=True)}")
# ## Plot 3D structures with PLAMS
images = {mol.properties.name: plams.view(mol, width=400) for mol in mols}
plams.plot_image_grid(images, rows=1, save_path="picture3.png")
# ## Plot 2D Lewis structures with RDKit
#
# The molecules can be aligned by using a benzene template. The ``regenerate`` option regenerates the molecule with RDkit to clean up the atomic positions.
draw_lewis_grid(mols, template_smiles=substrate_smiles, regenerate=True)