Nudged Elastic Band (NEB) for gasphase reaction with DFTB¶
Initial and final (approximate) states¶
By default, the NEB task performs geometry optimizations of the initial and final states before creating interpolated structures (images).
But it is a good idea to optimize the molecules before starting the NEB, to make sure that they are in reasonable positions.
Create the systems¶
Check out the GUI tutorial or the related “building structures” example to find out how to get the initial structures used here.
from scm.plams import view
from scm.base import ChemicalSystem
initial_sys = ChemicalSystem("""
System
Atoms
C 1.0065881512963437 -1.417955004240563 0.12885420257373528
C -0.08873018725926754 -2.1350000996605933 -0.03448649693154524
O -1.5081108856471217 1.002080375323799 -0.2045570136880532
N -0.4803377947492119 1.514182931120784 -0.052917508314934095
N 0.4992559331764258 2.001851603909277 0.09160944741412266
H 1.3565548327663348 -1.1143852514314225 1.1071131753303691
H 1.6148659535066119 -1.095826278535031 -0.7065218137603257
H -0.6985763255773102 -2.454234856430313 0.8007371044735704
H -0.4402537210929026 -2.435675244924285 -1.0129788309893883
End
End""")
view(initial_sys, width=150, height=150, direction="tilt_pca3", guess_bonds=True)
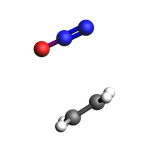
final_sys = ChemicalSystem("""
System
Atoms
C 0.8176518265162523 0.006185364744877703 0.11652129166388352
C -0.5531099061901967 -0.6363379504647356 -0.08529176454211394
O -1.4164729952124286 0.48607450912407413 -0.19677333330677277
N -0.6859030532374636 1.632537947036214 -0.08098563281192021
N 0.5074709667559635 1.4283821326486141 0.08689807666792455
H 1.2798721321383 -0.22791534308315498 1.0777663111036928
H 1.5305100901334066 -0.2099050616627963 -0.6819652771198405
H -0.866292408825807 -1.2485848049850283 0.7685478972017538
H -0.6137266520780276 -1.2304367933580558 -1.0047175688566132
End
End""")
view(final_sys, width=150, height=150, direction="tilt_pca3", guess_bonds=True)
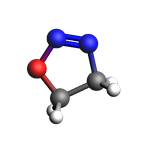
NEB and DFTB settings. Run the Job.¶
from scm.plams import Settings, AMSJob
settings = Settings()
# Input options for the AMS driver
settings.input.ams.Task = "NEB"
settings.input.ams.Properties.PESPointCharacter = "Yes"
# Input options for the engine (DFTB in this case)
settings.input.DFTB.Model = "GFN1-xTB"
# You can pass multiple systems to an AMSJob by using a dictionary of molecules.
# The key of the dictionary will be used as the header of the 'System' block
molecules = {"": initial_sys, "final": final_sys}
# Create and run the job
job = AMSJob(settings=settings, molecule=molecules, name="NEB")
print(job.get_input())
Properties
PESPointCharacter Yes
End
Task NEB
System
Atoms
C 1.0065881512963437 -1.417955004240563 0.12885420257373528
C -0.08873018725926754 -2.1350000996605933 -0.03448649693154524
O -1.5081108856471217 1.002080375323799 -0.2045570136880532
N -0.4803377947492119 1.514182931120784 -0.052917508314934095
N 0.4992559331764258 2.001851603909277 0.09160944741412266
H 1.3565548327663348 -1.1143852514314225 1.1071131753303691
H 1.6148659535066119 -1.095826278535031 -0.7065218137603257
... output trimmed ....
O -1.4164729952124286 0.48607450912407413 -0.19677333330677277
N -0.6859030532374636 1.632537947036214 -0.08098563281192021
N 0.5074709667559635 1.4283821326486141 0.08689807666792455
H 1.2798721321383 -0.22791534308315498 1.0777663111036928
H 1.5305100901334066 -0.2099050616627963 -0.6819652771198405
H -0.866292408825807 -1.2485848049850283 0.7685478972017538
H -0.6137266520780276 -1.2304367933580558 -1.0047175688566132
End
End
Engine DFTB
Model GFN1-xTB
EndEngine
job.run();
[28.02|09:45:50] JOB NEB STARTED
[28.02|09:45:50] JOB NEB RUNNING
[28.02|09:45:51] JOB NEB FINISHED
[28.02|09:45:51] JOB NEB SUCCESSFUL
# job = AMSJob.load_external("/path/to/ams.rkf") # if loading results from disk
assert job.ok(), "Looks like NEB calculation failed?"
print("Successful NEB calculation!")
Successful NEB calculation!
NEB Results¶
print(job.results.engine_names())
['NEB_TS_final']
from pprint import pprint
pes_point_character = job.results.readrkf("AMSResults", "PESPointCharacter", file="NEB_TS_final")
print(f"{pes_point_character=}")
pes_point_character='transition state'
neb_res = job.results.get_neb_results()
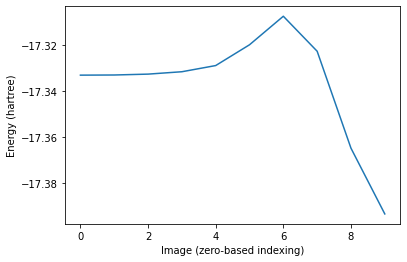
pprint(neb_res) # energies in hartree
{'Climbing': True,
'Energies': [-17.333016762960952,
-17.33294947958635,
-17.33256809516803,
-17.331527201901654,
-17.328829238015103,
-17.319753903625962,
-17.307297193368917,
-17.322575012605878,
-17.364900430989824,
-17.393606196413522],
'HighestIndex': 6,
'HistoryIndices': [309, 301, 302, 303, 304, 305, 306, 307, 308, 310],
'LeftBarrier': 0.025719569592034475,
'Molecules': [Molecule('C2H4N2O' at 0x7e3cea270370),
Molecule('C2H4N2O' at 0x7e3cea270670),
Molecule('C2H4N2O' at 0x7e3cea270790),
Molecule('C2H4N2O' at 0x7e3cea270280),
Molecule('C2H4N2O' at 0x7e3cea270b20),
Molecule('C2H4N2O' at 0x7e3cea270dc0),
Molecule('C2H4N2O' at 0x7e3cea6c9160),
Molecule('C2H4N2O' at 0x7e3cf20742b0),
Molecule('C2H4N2O' at 0x7e3ce9ffb0a0),
Molecule('C2H4N2O' at 0x7e3ce9ffb310)],
'ReactionEnergy': -0.06058943345257006,
'RightBarrier': 0.08630900304460454,
'nImages': 8,
'nIterations': 216}
import matplotlib.pyplot as plt
neb_res = job.results.get_neb_results()
fig, ax = plt.subplots()
ax.plot(neb_res["Energies"])
ax.set_ylabel("Energy (hartree)")
ax.set_xlabel("Image (zero-based indexing)");
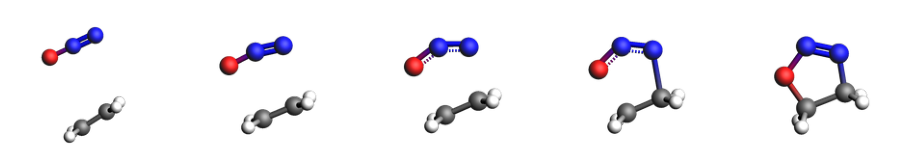
import matplotlib.pyplot as plt
from scm.plams import view
neb_res = job.results.get_neb_results()
mols = neb_res["Molecules"] # these are PLAMS Molecule, not ChemicalSystem
indices = [0, 5, 6, 7, 9] # show only these
fig, axes = plt.subplots(1, len(indices), figsize=(16, 16))
for ax, i in zip(axes, indices):
img = view(mols[i], width=160, height=160, direction="tilt_pca3", guess_bonds=True)
ax.imshow(img)
ax.axis("off")
axes;
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial and final (approximate) states
#
# By default, the NEB task performs geometry optimizations of the initial and final states before creating interpolated structures (images).
#
# But it is a good idea to optimize the molecules before starting the NEB, to make sure that they are in reasonable positions.
#
# ## Create the systems
#
# Check out the GUI tutorial or the related "building structures" example to find out how to get the initial structures used here.
from scm.plams import view
from scm.base import ChemicalSystem
initial_sys = ChemicalSystem(
"""
System
Atoms
C 1.0065881512963437 -1.417955004240563 0.12885420257373528
C -0.08873018725926754 -2.1350000996605933 -0.03448649693154524
O -1.5081108856471217 1.002080375323799 -0.2045570136880532
N -0.4803377947492119 1.514182931120784 -0.052917508314934095
N 0.4992559331764258 2.001851603909277 0.09160944741412266
H 1.3565548327663348 -1.1143852514314225 1.1071131753303691
H 1.6148659535066119 -1.095826278535031 -0.7065218137603257
H -0.6985763255773102 -2.454234856430313 0.8007371044735704
H -0.4402537210929026 -2.435675244924285 -1.0129788309893883
End
End"""
)
view(initial_sys, width=150, height=150, direction="tilt_pca3", guess_bonds=True, picture_path="picture1.png")
final_sys = ChemicalSystem(
"""
System
Atoms
C 0.8176518265162523 0.006185364744877703 0.11652129166388352
C -0.5531099061901967 -0.6363379504647356 -0.08529176454211394
O -1.4164729952124286 0.48607450912407413 -0.19677333330677277
N -0.6859030532374636 1.632537947036214 -0.08098563281192021
N 0.5074709667559635 1.4283821326486141 0.08689807666792455
H 1.2798721321383 -0.22791534308315498 1.0777663111036928
H 1.5305100901334066 -0.2099050616627963 -0.6819652771198405
H -0.866292408825807 -1.2485848049850283 0.7685478972017538
H -0.6137266520780276 -1.2304367933580558 -1.0047175688566132
End
End"""
)
view(final_sys, width=150, height=150, direction="tilt_pca3", guess_bonds=True, picture_path="picture2.png")
# ## NEB and DFTB settings. Run the Job.
from scm.plams import Settings, AMSJob
settings = Settings()
# Input options for the AMS driver
settings.input.ams.Task = "NEB"
settings.input.ams.Properties.PESPointCharacter = "Yes"
# Input options for the engine (DFTB in this case)
settings.input.DFTB.Model = "GFN1-xTB"
# You can pass multiple systems to an AMSJob by using a dictionary of molecules.
# The key of the dictionary will be used as the header of the 'System' block
molecules = {"": initial_sys, "final": final_sys}
# Create and run the job
job = AMSJob(settings=settings, molecule=molecules, name="NEB")
print(job.get_input())
job.run()
# job = AMSJob.load_external("/path/to/ams.rkf") # if loading results from disk
assert job.ok(), "Looks like NEB calculation failed?"
print("Successful NEB calculation!")
# ## NEB Results
print(job.results.engine_names())
from pprint import pprint
pes_point_character = job.results.readrkf("AMSResults", "PESPointCharacter", file="NEB_TS_final")
print(f"{pes_point_character=}")
neb_res = job.results.get_neb_results()
pprint(neb_res) # energies in hartree
import matplotlib.pyplot as plt
neb_res = job.results.get_neb_results()
fig, ax = plt.subplots()
ax.plot(neb_res["Energies"])
ax.set_ylabel("Energy (hartree)")
ax.set_xlabel("Image (zero-based indexing)")
import matplotlib.pyplot as plt
from scm.plams import view
neb_res = job.results.get_neb_results()
mols = neb_res["Molecules"] # these are PLAMS Molecule, not ChemicalSystem
indices = [0, 5, 6, 7, 9] # show only these
fig, axes = plt.subplots(1, len(indices), figsize=(16, 16))
for ax, i in zip(axes, indices):
img = view(mols[i], width=160, height=160, direction="tilt_pca3", guess_bonds=True)
ax.imshow(img)
ax.axis("off")
axes