Molecule and Bond Counts from Reactive MD¶
Run a short reactive molecular dynamics simulation with ReaxFF and tabulate the time evolution of molecule and bond-type counts from the generated trajectory.
Molecule and Bond Counts from Reactive MD¶
This example performs molecule and bond counting from a reactive MD trajectory. It first runs a short ReaxFF simulation, then tabulates and plots counts per frame from the generated ams.rkf.
The MD run requires a ReaxFF license. If you already have a reactive MD trajectory with molecule data, you can skip the MD cell and set ams_rkf_path to your own file.
Initial Imports¶
from collections import Counter
from pathlib import Path
from typing import Dict, List, Literal, Optional, Sequence, Set, Tuple
import matplotlib.pyplot as plt
from matplotlib.axes import Axes
from scm.plams import AMSJob, Molecule, Settings, Trajectory, from_smiles, packmol, view
Run a short reactive MD simulation¶
This produces an ams.rkf trajectory with molecule and bond information for analysis.
o2 = from_smiles("O=O")
h2 = from_smiles("[HH]")
mixture = packmol(molecules=[o2, h2], n_molecules=[4, 4], density=1.0)
view(mixture, direction="tilt_z", width=300, height=300)
md_settings = Settings()
md_settings.input.reaxff.forcefield = "CHO.ff"
md_settings.input.ams.task = "MolecularDynamics"
md_settings.input.ams.MolecularDynamics.NSteps = 3000
md_settings.input.ams.MolecularDynamics.TimeStep = 0.5
md_settings.input.ams.MolecularDynamics.InitialVelocities.Temperature = 3500
md_settings.input.ams.MolecularDynamics.Trajectory.WriteMolecules = "Yes"
md_settings.input.ams.MolecularDynamics.Trajectory.WriteBonds = "Yes"
md_settings.runscript.nproc = 1
md_settings.runscript.preamble_lines = ["export OMP_NUM_THREADS=1"]
md_job = AMSJob(name="reactive_md_toy", settings=md_settings, molecule=mixture)
md_results = md_job.run();
[30.03|16:56:24] JOB reactive_md_toy STARTED
[30.03|16:56:24] JOB reactive_md_toy RUNNING
[30.03|16:56:26] JOB reactive_md_toy FINISHED
[30.03|16:56:26] JOB reactive_md_toy SUCCESSFUL
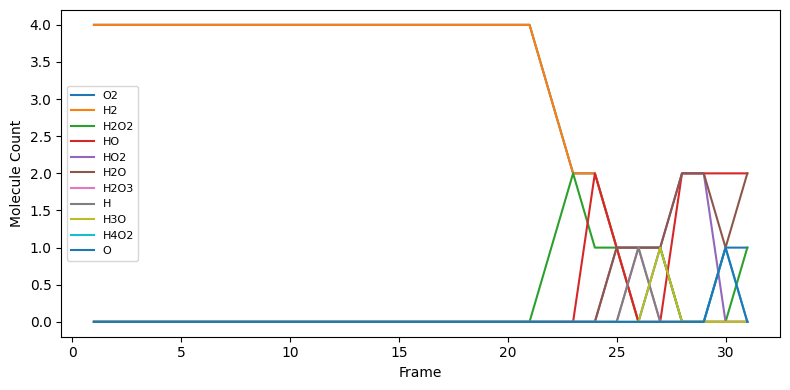
Molecule counts per frame¶
For each frame, count how many molecules of each detected species are present.
import pandas as pd
n_molecules = int(md_job.results.readrkf("Molecules", "Num molecules"))
molecule_type_ids = list(range(1, n_molecules + 1))
molecule_names = [
md_job.results.readrkf("Molecules", f"Molecule name {i}") for i in molecule_type_ids
]
mols_type_history = md_job.results.get_history_property("Mols.Type")
molecule_counts = []
frame = 1
for mol_types in mols_type_history:
counter = Counter(mol_types)
counts = {"Frame": frame}
for i in molecule_type_ids:
counts[molecule_names[i - 1]] = counter[i]
molecule_counts.append(counts)
frame += 1
molecule_counts_df = pd.DataFrame(molecule_counts)
molecule_counts_df
Frame | O2 | H2 | H2O2 | HO | HO2 | H2O | H2O3 | H | H3O | H4O2 | O | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
0 | 1 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
1 | 2 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
2 | 3 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
3 | 4 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
4 | 5 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 | 6 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
6 | 7 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
7 | 8 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
8 | 9 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
9 | 10 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
10 | 11 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
11 | 12 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
12 | 13 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
13 | 14 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
14 | 15 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
15 | 16 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
16 | 17 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
17 | 18 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
18 | 19 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
19 | 20 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
20 | 21 | 4 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
21 | 22 | 3 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
22 | 23 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
23 | 24 | 2 | 2 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
24 | 25 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
25 | 26 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 |
26 | 27 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
27 | 28 | 0 | 0 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
28 | 29 | 0 | 0 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
29 | 30 | 1 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 1 |
30 | 31 | 1 | 0 | 1 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
fig, ax = plt.subplots(figsize=(8, 4))
x = molecule_counts_df["Frame"]
for i, name in enumerate(molecule_names):
y = molecule_counts_df[name]
ax.plot(x, y, label=name)
ax.set_xlabel("Frame")
ax.set_ylabel("Molecule Count")
ax.legend(loc="best", fontsize=8)
fig.tight_layout()
ax;
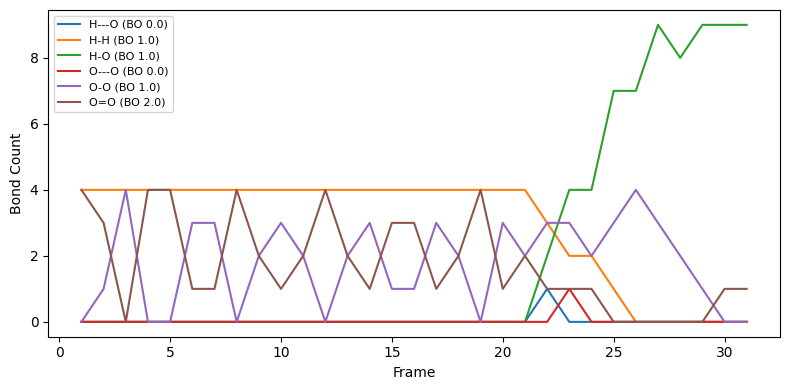
Bond counts per frame¶
Count bond types per frame, using bond order rounded to either integer or half-integer values.
trajectory = Trajectory(md_job.results.rkfpath())
n_frames = len(trajectory)
def bo_to_symbol(bo):
s = "---"
if bo == 0.5:
s = "--"
if bo == 1.0:
s = "-"
if bo == 1.5:
s = "=="
if bo == 2.0:
s = "="
if bo == 2.5:
s = "≡≡"
if bo == 3.0:
s = "≡"
return s
all_bonds = {}
for frame_index, mol in enumerate(trajectory):
frame_counts: Counter = Counter()
seen_pairs: Set[Tuple[int, int]] = set()
for bond in mol.bonds:
pair = tuple(sorted((mol.index(bond.atom1), mol.index(bond.atom2))))
if pair in seen_pairs:
continue
seen_pairs.add(pair)
symbols = sorted((bond.atom1.symbol, bond.atom2.symbol))
bo = float(round(bond.order))
label = f"{symbols[0]}{bo_to_symbol(bo)}{symbols[1]} (BO {bo:.1f})"
frame_counts[label] += 1
for label, count in frame_counts.items():
if label not in all_bonds:
all_bonds[label] = [0] * n_frames
all_bonds[label][frame_index] = int(count)
bond_names = sorted(all_bonds.keys())
bond_counts = []
for frame_index in range(n_frames):
counts = {"Frame": frame_index + 1}
for name in bond_names:
counts[name] = all_bonds[name][frame_index]
bond_counts.append(counts)
bond_counts_df = pd.DataFrame(bond_counts)
bond_counts_df
Frame | H—O (BO 0.0) | H-H (BO 1.0) | H-O (BO 1.0) | O—O (BO 0.0) | O-O (BO 1.0) | O=O (BO 2.0) | |
|---|---|---|---|---|---|---|---|
0 | 1 | 0 | 4 | 0 | 0 | 0 | 4 |
1 | 2 | 0 | 4 | 0 | 0 | 1 | 3 |
2 | 3 | 0 | 4 | 0 | 0 | 4 | 0 |
3 | 4 | 0 | 4 | 0 | 0 | 0 | 4 |
4 | 5 | 0 | 4 | 0 | 0 | 0 | 4 |
5 | 6 | 0 | 4 | 0 | 0 | 3 | 1 |
6 | 7 | 0 | 4 | 0 | 0 | 3 | 1 |
7 | 8 | 0 | 4 | 0 | 0 | 0 | 4 |
8 | 9 | 0 | 4 | 0 | 0 | 2 | 2 |
9 | 10 | 0 | 4 | 0 | 0 | 3 | 1 |
10 | 11 | 0 | 4 | 0 | 0 | 2 | 2 |
11 | 12 | 0 | 4 | 0 | 0 | 0 | 4 |
12 | 13 | 0 | 4 | 0 | 0 | 2 | 2 |
13 | 14 | 0 | 4 | 0 | 0 | 3 | 1 |
14 | 15 | 0 | 4 | 0 | 0 | 1 | 3 |
15 | 16 | 0 | 4 | 0 | 0 | 1 | 3 |
16 | 17 | 0 | 4 | 0 | 0 | 3 | 1 |
17 | 18 | 0 | 4 | 0 | 0 | 2 | 2 |
18 | 19 | 0 | 4 | 0 | 0 | 0 | 4 |
19 | 20 | 0 | 4 | 0 | 0 | 3 | 1 |
20 | 21 | 0 | 4 | 0 | 0 | 2 | 2 |
21 | 22 | 1 | 3 | 2 | 0 | 3 | 1 |
22 | 23 | 0 | 2 | 4 | 1 | 3 | 1 |
23 | 24 | 0 | 2 | 4 | 0 | 2 | 1 |
24 | 25 | 0 | 1 | 7 | 0 | 3 | 0 |
25 | 26 | 0 | 0 | 7 | 0 | 4 | 0 |
26 | 27 | 0 | 0 | 9 | 0 | 3 | 0 |
27 | 28 | 0 | 0 | 8 | 0 | 2 | 0 |
28 | 29 | 0 | 0 | 9 | 0 | 1 | 0 |
29 | 30 | 0 | 0 | 9 | 0 | 0 | 1 |
30 | 31 | 0 | 0 | 9 | 0 | 0 | 1 |
fig, ax = plt.subplots(figsize=(8, 4))
x = bond_counts_df["Frame"]
for i, name in enumerate(bond_names):
y = bond_counts_df[name]
ax.plot(x, y, label=name)
ax.set_xlabel("Frame")
ax.set_ylabel("Bond Count")
ax.legend(loc="best", fontsize=8)
fig.tight_layout()
ax;
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Molecule and Bond Counts from Reactive MD
#
# This example performs molecule and bond counting from a reactive MD trajectory. It first runs a short ReaxFF simulation, then tabulates and plots counts per frame from the generated `ams.rkf`.
# The MD run requires a ReaxFF license. If you already have a reactive MD trajectory with molecule data, you can skip the MD cell and set `ams_rkf_path` to your own file.
#
# ### Initial Imports
from collections import Counter
from pathlib import Path
from typing import Dict, List, Literal, Optional, Sequence, Set, Tuple
import matplotlib.pyplot as plt
from matplotlib.axes import Axes
from scm.plams import AMSJob, Molecule, Settings, Trajectory, from_smiles, packmol, view
# ### Run a short reactive MD simulation
#
# This produces an `ams.rkf` trajectory with molecule and bond information for analysis.
#
o2 = from_smiles("O=O")
h2 = from_smiles("[HH]")
mixture = packmol(molecules=[o2, h2], n_molecules=[4, 4], density=1.0)
view(mixture, direction="tilt_z", width=300, height=300)
md_settings = Settings()
md_settings.input.reaxff.forcefield = "CHO.ff"
md_settings.input.ams.task = "MolecularDynamics"
md_settings.input.ams.MolecularDynamics.NSteps = 3000
md_settings.input.ams.MolecularDynamics.TimeStep = 0.5
md_settings.input.ams.MolecularDynamics.InitialVelocities.Temperature = 3500
md_settings.input.ams.MolecularDynamics.Trajectory.WriteMolecules = "Yes"
md_settings.input.ams.MolecularDynamics.Trajectory.WriteBonds = "Yes"
md_settings.runscript.nproc = 1
md_settings.runscript.preamble_lines = ["export OMP_NUM_THREADS=1"]
md_job = AMSJob(name="reactive_md_toy", settings=md_settings, molecule=mixture)
md_results = md_job.run()
# ### Molecule counts per frame
#
# For each frame, count how many molecules of each detected species are present.
#
import pandas as pd
n_molecules = int(md_job.results.readrkf("Molecules", "Num molecules"))
molecule_type_ids = list(range(1, n_molecules + 1))
molecule_names = [md_job.results.readrkf("Molecules", f"Molecule name {i}") for i in molecule_type_ids]
mols_type_history = md_job.results.get_history_property("Mols.Type")
molecule_counts = []
frame = 1
for mol_types in mols_type_history:
counter = Counter(mol_types)
counts = {"Frame": frame}
for i in molecule_type_ids:
counts[molecule_names[i - 1]] = counter[i]
molecule_counts.append(counts)
frame += 1
molecule_counts_df = pd.DataFrame(molecule_counts)
print(molecule_counts_df)
fig, ax = plt.subplots(figsize=(8, 4))
x = molecule_counts_df["Frame"]
for i, name in enumerate(molecule_names):
y = molecule_counts_df[name]
ax.plot(x, y, label=name)
ax.set_xlabel("Frame")
ax.set_ylabel("Molecule Count")
ax.legend(loc="best", fontsize=8)
fig.tight_layout()
ax
ax.figure.savefig("picture1.png")
# ## Bond counts per frame
#
# Count bond types per frame, using bond order rounded to either integer or half-integer values.
#
trajectory = Trajectory(md_job.results.rkfpath())
n_frames = len(trajectory)
def bo_to_symbol(bo):
s = "---"
if bo == 0.5:
s = "--"
if bo == 1.0:
s = "-"
if bo == 1.5:
s = "=="
if bo == 2.0:
s = "="
if bo == 2.5:
s = "≡≡"
if bo == 3.0:
s = "≡"
return s
all_bonds = {}
for frame_index, mol in enumerate(trajectory):
frame_counts: Counter = Counter()
seen_pairs: Set[Tuple[int, int]] = set()
for bond in mol.bonds:
pair = tuple(sorted((mol.index(bond.atom1), mol.index(bond.atom2))))
if pair in seen_pairs:
continue
seen_pairs.add(pair)
symbols = sorted((bond.atom1.symbol, bond.atom2.symbol))
bo = float(round(bond.order))
label = f"{symbols[0]}{bo_to_symbol(bo)}{symbols[1]} (BO {bo:.1f})"
frame_counts[label] += 1
for label, count in frame_counts.items():
if label not in all_bonds:
all_bonds[label] = [0] * n_frames
all_bonds[label][frame_index] = int(count)
bond_names = sorted(all_bonds.keys())
bond_counts = []
for frame_index in range(n_frames):
counts = {"Frame": frame_index + 1}
for name in bond_names:
counts[name] = all_bonds[name][frame_index]
bond_counts.append(counts)
bond_counts_df = pd.DataFrame(bond_counts)
print(bond_counts_df)
fig, ax = plt.subplots(figsize=(8, 4))
x = bond_counts_df["Frame"]
for i, name in enumerate(bond_names):
y = bond_counts_df[name]
ax.plot(x, y, label=name)
ax.set_xlabel("Frame")
ax.set_ylabel("Bond Count")
ax.legend(loc="best", fontsize=8)
fig.tight_layout()
ax
ax.figure.savefig("picture2.png")