Simulating XRD Patterns from a CIF Structure¶
Convert a structure between ASE, CIF, and pymatgen representations and calculate an X-ray diffraction pattern from the resulting crystal model.
Initial imports¶
from scm.plams import *
from ase import Atoms
from pymatgen.core.structure import Structure
from pymatgen.analysis.diffraction.xrd import XRDCalculator
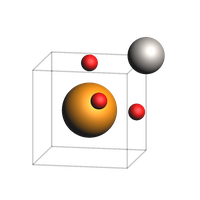
Create ASE atoms object for BaTiO3¶
at = Atoms(
symbols=[
"Ba",
"Ti",
"O",
"O",
"O",
],
scaled_positions=[
[
0.0,
0.0,
0.0,
],
[0.5, 0.5, 0.5],
[0.0, 0.0, 0.5],
[0.0, 0.5, 0.0],
[0.5, 0.0, 0.0],
],
cell=[4.01, 4.01, 4.01],
pbc=(True, True, True),
)
view(fromASE(at), fixed_atom_size=False, direction="large_tilt_z", width=200, height=200)
Save ASE Atoms to .cif format¶
fname = "batio3.cif"
at.write(fname)
Load .cif in pymatgen and calculate XRD¶
Available radiation sources:
print(f"Available radiation sources: {XRDCalculator.AVAILABLE_RADIATION}")
Available radiation sources: ('CuKa', 'CuKa2', 'CuKa1', 'CuKb1', 'MoKa', 'MoKa2', 'MoKa1', 'MoKb1', 'CrKa', 'CrKa2', 'CrKa1', 'CrKb1', 'FeKa', 'FeKa2', 'FeKa1', 'FeKb1', 'CoKa', 'CoKa2', 'CoKa1', 'CoKb1', 'AgKa', 'AgKa2', 'AgKa1', 'AgKb1')
Let’s choose Cu K-alpha (default):
structure = Structure.from_file(fname)
xrd_calc = XRDCalculator(wavelength="CuKa")
xrd_calc.show_plot(structure)
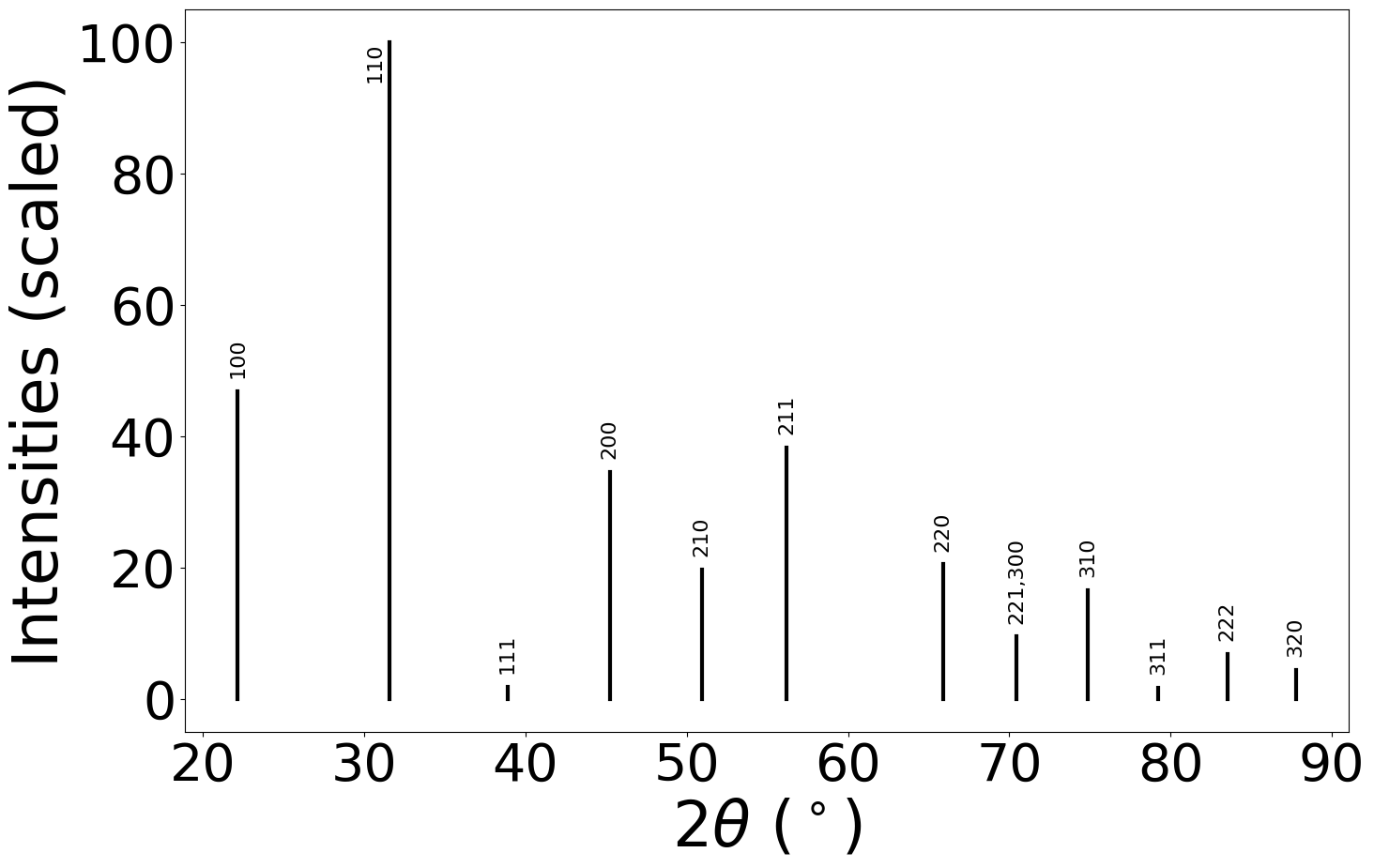
pattern = xrd_calc.get_pattern(structure)
print("2*Theta Intensity hkl d_hkl(angstrom)")
for two_theta, intensity, hkls, d_hkl in zip(pattern.x, pattern.y, pattern.hkls, pattern.d_hkls):
hkl_tuples = [hkl["hkl"] for hkl in hkls]
for hkl in hkl_tuples:
label = ", ".join(map(str, hkl))
print(f"{two_theta:.2f} {intensity:.2f} {hkl} {d_hkl:.3f}")
2*Theta Intensity hkl d_hkl(angstrom)
22.17 46.84 (1, 0, 0) 4.010
31.55 100.00 (1, 1, 0) 2.835
38.90 1.83 (1, 1, 1) 2.315
45.23 34.58 (2, 0, 0) 2.005
50.92 19.69 (2, 1, 0) 1.793
56.19 38.27 (2, 1, 1) 1.637
65.88 20.48 (2, 2, 0) 1.418
70.44 9.47 (2, 2, 1) 1.337
70.44 9.47 (3, 0, 0) 1.337
74.88 16.60 (3, 1, 0) 1.268
79.23 1.68 (3, 1, 1) 1.209
83.51 6.82 (2, 2, 2) 1.158
87.76 4.44 (3, 2, 0) 1.112
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Initial imports
from scm.plams import *
from ase import Atoms
from pymatgen.core.structure import Structure
from pymatgen.analysis.diffraction.xrd import XRDCalculator
# ## Create ASE atoms object for BaTiO3
at = Atoms(
symbols=[
"Ba",
"Ti",
"O",
"O",
"O",
],
scaled_positions=[
[
0.0,
0.0,
0.0,
],
[0.5, 0.5, 0.5],
[0.0, 0.0, 0.5],
[0.0, 0.5, 0.0],
[0.5, 0.0, 0.0],
],
cell=[4.01, 4.01, 4.01],
pbc=(True, True, True),
)
view(fromASE(at), fixed_atom_size=False, direction="large_tilt_z", width=200, height=200, picture_path="picture1.png")
# ## Save ASE Atoms to .cif format
fname = "batio3.cif"
at.write(fname)
# ## Load .cif in pymatgen and calculate XRD
# Available radiation sources:
print(f"Available radiation sources: {XRDCalculator.AVAILABLE_RADIATION}")
# Let's choose Cu K-alpha (default):
structure = Structure.from_file(fname)
xrd_calc = XRDCalculator(wavelength="CuKa")
xrd_calc.show_plot(structure)
pattern = xrd_calc.get_pattern(structure)
print("2*Theta Intensity hkl d_hkl(angstrom)")
for two_theta, intensity, hkls, d_hkl in zip(pattern.x, pattern.y, pattern.hkls, pattern.d_hkls):
hkl_tuples = [hkl["hkl"] for hkl in hkls]
for hkl in hkl_tuples:
label = ", ".join(map(str, hkl))
print(f"{two_theta:.2f} {intensity:.2f} {hkl} {d_hkl:.3f}")