Surface Energy with ReaxFF¶
Here we calculate the surface energy of ZnO(10-10).
Optimize the bulk the structure
Cut out slabs with progressively bigger thickness
Optimize the slabs with ReaxFF
Plot energy vs. number of atoms and fit a straight line
Convert the y-intercept of the straight line to the surface energy
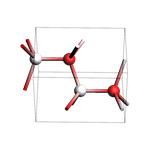
Bulk wurtzite ZnO¶
from scm.base import ChemicalSystem
from scm.plams import view
bulk = ChemicalSystem(
"""
System
Atoms
Zn 1.64483022629693 0.94964317652279 0.00064920490187
Zn 1.64483022629108 -0.94964317650531 2.68804611338770
O 1.64483022629670 0.94964317652865 2.01489847647198
O 1.64483022629084 -0.94964317649945 4.70229538495781
End
Lattice
1.64483022628585 -2.84892951942711 -0.00000000000866
1.64483022630247 2.84892951943670 0.00000000000826
-0.00000000000063 0.00000000001572 5.37479381698294
End
End
"""
)
view(bulk, width=150, height=150, direction="tilt_x", guess_bonds=True)
[10.04|12:54:49] Starting Xvfb...
[10.04|12:54:50] Xvfb started
Lattice optimization with ReaxFF¶
import scm.plams as plams
engine_s = plams.Settings()
engine_s.input.ReaxFF.ForceField = "ZnOH.ff"
engine_s.runscript.nproc = 1
s = plams.Settings()
s.input.ams.Task = "GeometryOptimization"
s.input.ams.GeometryOptimization.OptimizeLattice = "Yes"
s.input.ams.GeometryOptimization.Convergence.Quality = "Good"
s += engine_s.copy()
job = plams.AMSJob(settings=s, molecule=bulk, name="bulk_opt")
job.run();
[10.04|12:54:51] JOB bulk_opt STARTED
[10.04|12:54:51] JOB bulk_opt RUNNING
[10.04|12:54:51] JOB bulk_opt FINISHED
[10.04|12:54:51] JOB bulk_opt SUCCESSFUL
optimized_bulk = job.results.get_main_system()
optimized_bulk.symmetrize_cell()
print("Lengths (ang.): ", optimized_bulk.lattice.get_lengths())
print("Angles (deg.): ", optimized_bulk.lattice.get_angles(unit="degree"))
Lengths (ang.): [3.28000889 3.28000889 5.31353255]
Angles (deg.): [ 90. 90. 120.]
Create slabs¶
num_layers_list = [6, 8, 10, 12, 14]
miller = (1, 0, -1, 0)
slabs = [optimized_bulk.make_slab_layers(miller=miller, num_layers=x) for x in num_layers_list]
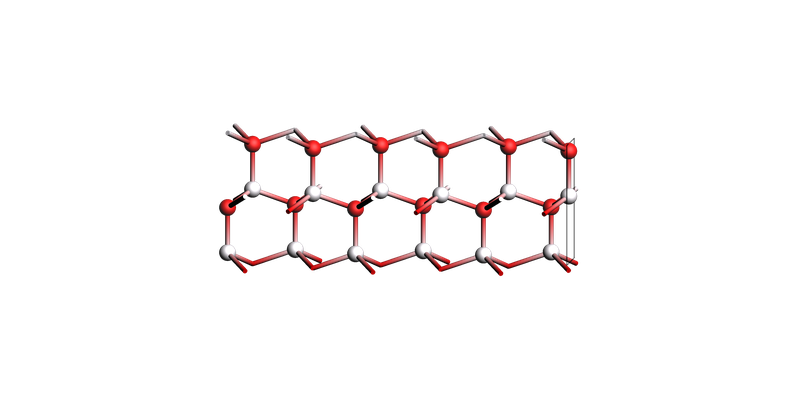
The thinnest slab (shown horizontally):
view(slabs[0], direction="tilt_x", guess_bonds=True)
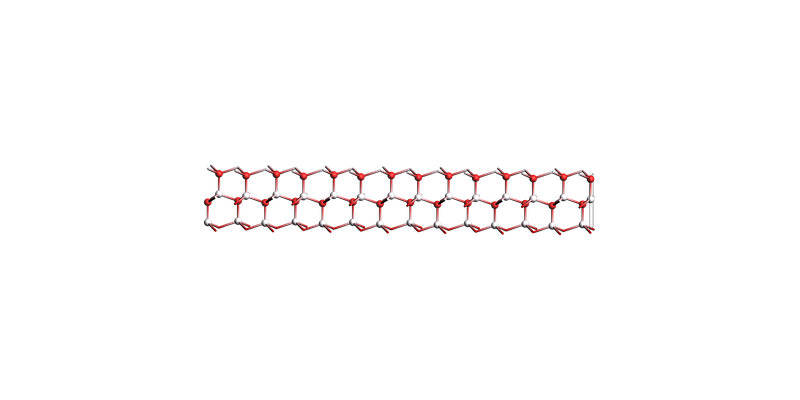
The thickest slab:
view(slabs[-1], direction="tilt_x", guess_bonds=True)
Run slab geometry optimizations¶
from scm.plams import AMSJob
ss = plams.Settings()
ss.input.ams.Task = "GeometryOptimization"
ss += engine_s.copy()
slab_jobs = []
for num_layers, slab in zip(num_layers_list, slabs):
millerstring = "".join(str(x) for x in miller)
name = f"{num_layers}layers_{millerstring}"
job = AMSJob(settings=ss.copy(), molecule=slab, name=name)
slab_jobs.append(job)
for job in slab_jobs:
job.run()
[10.04|12:54:53] JOB 6layers_10-10 STARTED
[10.04|12:54:53] JOB 6layers_10-10 RUNNING
[10.04|12:54:54] JOB 6layers_10-10 FINISHED
[10.04|12:54:54] JOB 6layers_10-10 SUCCESSFUL
[10.04|12:54:54] JOB 8layers_10-10 STARTED
... output trimmed ....
[10.04|12:54:54] JOB 12layers_10-10 SUCCESSFUL
[10.04|12:54:54] JOB 14layers_10-10 STARTED
[10.04|12:54:54] JOB 14layers_10-10 RUNNING
[10.04|12:54:54] JOB 14layers_10-10 FINISHED
[10.04|12:54:54] JOB 14layers_10-10 SUCCESSFUL
import matplotlib.pyplot as plt
import numpy as np
def getresults(job):
main_sys = job.results.get_main_system()
n_atoms = len(main_sys)
energy = job.results.get_energy(unit="hartree")
vecs = main_sys.lattice.vectors
area = np.linalg.norm(np.cross(vecs[0], vecs[1]))
return n_atoms, energy, area
data = sorted(getresults(job) for job in slab_jobs) # sorts on number of atoms
x, y, areas = zip(*data) # x = number of atoms, y = energy
fig, ax = plt.subplots()
ax.plot(x, y, ".")
ax.set_xlim(0, max(x) + 1)
ax.set_ylabel("Energy (hartree)")
ax.set_xlabel("Number of atoms")
ax;
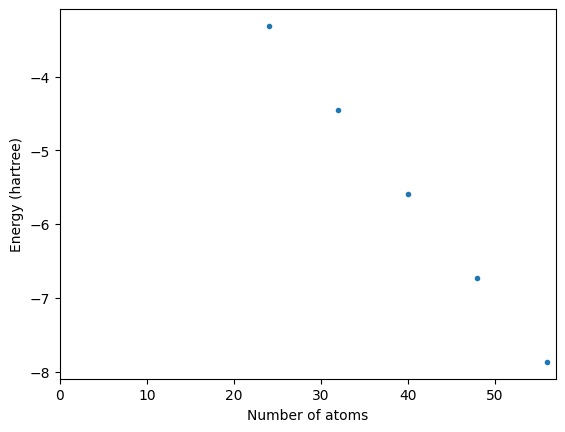
We need to fit a straight line through the points and see where it intercepts 0 on the x axis.
Fit a straight line and extrapolate to 0 atoms (pure “surface”)¶
from scipy.stats import linregress
from scm.base import Units
k = 3 # use the last k points
surfacearea = areas[-1] # all should be identical; just use the last number
result = linregress(x[-k:], y[-k:])
lineminx, lineminy = 0, result.intercept
linemaxx, linemaxy = max(x), result.slope * max(x) + result.intercept
fig, ax = plt.subplots()
ax.plot(x, y, ".")
ax.plot([lineminx, linemaxx], [lineminy, linemaxy], "--")
ax.set_xlim(0, max(x) + 1)
ax.set_ylabel("Energy (hartree)")
ax.set_xlabel("Number of atoms")
ax;
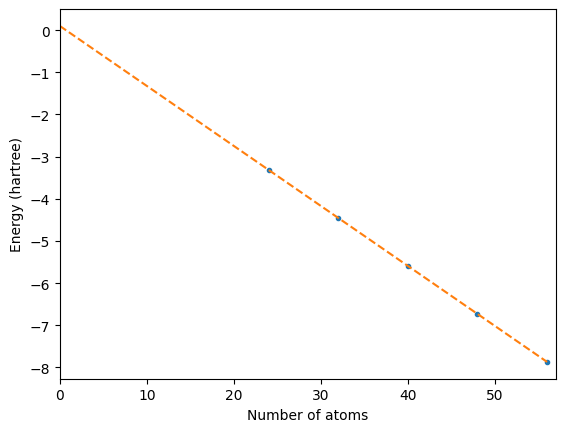
Extract surface energy from y-axis intercept¶
hartree2J = Units.conversion_factor("hartree", "J")
angstrom2m = Units.conversion_factor("angstrom", "m")
conversion = hartree2J / angstrom2m**2
Esurf = conversion * result.intercept / (2 * surfacearea)
print(f"Intercept = {result.intercept:.6f} hartree")
print(f"Surface energy: {Esurf:.3f} J/m^2")
Intercept = 0.100491 hartree
Surface energy: 1.257 J/m^2
Final notes on surface energy¶
In the calculation of the surface energy, we never used the energy of the bulk! This is best practice.
The surface energy is expressed in J/m^2; the area of the unit cell will depend strongly on the optimized the lattice parameters.
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## Bulk wurtzite ZnO
from scm.base import ChemicalSystem
from scm.plams import view
bulk = ChemicalSystem(
"""
System
Atoms
Zn 1.64483022629693 0.94964317652279 0.00064920490187
Zn 1.64483022629108 -0.94964317650531 2.68804611338770
O 1.64483022629670 0.94964317652865 2.01489847647198
O 1.64483022629084 -0.94964317649945 4.70229538495781
End
Lattice
1.64483022628585 -2.84892951942711 -0.00000000000866
1.64483022630247 2.84892951943670 0.00000000000826
-0.00000000000063 0.00000000001572 5.37479381698294
End
End
"""
)
view(bulk, width=150, height=150, direction="tilt_x", guess_bonds=True, picture_path="picture1.png")
# ## Lattice optimization with ReaxFF
import scm.plams as plams
engine_s = plams.Settings()
engine_s.input.ReaxFF.ForceField = "ZnOH.ff"
engine_s.runscript.nproc = 1
s = plams.Settings()
s.input.ams.Task = "GeometryOptimization"
s.input.ams.GeometryOptimization.OptimizeLattice = "Yes"
s.input.ams.GeometryOptimization.Convergence.Quality = "Good"
s += engine_s.copy()
job = plams.AMSJob(settings=s, molecule=bulk, name="bulk_opt")
job.run()
optimized_bulk = job.results.get_main_system()
optimized_bulk.symmetrize_cell()
print("Lengths (ang.): ", optimized_bulk.lattice.get_lengths())
print("Angles (deg.): ", optimized_bulk.lattice.get_angles(unit="degree"))
# ## Create slabs
num_layers_list = [6, 8, 10, 12, 14]
miller = (1, 0, -1, 0)
slabs = [optimized_bulk.make_slab_layers(miller=miller, num_layers=x) for x in num_layers_list]
# The thinnest slab (shown horizontally):
view(slabs[0], direction="tilt_x", guess_bonds=True, picture_path="picture2.png")
# The thickest slab:
view(slabs[-1], direction="tilt_x", guess_bonds=True, picture_path="picture3.png")
# ## Run slab geometry optimizations
from scm.plams import AMSJob
ss = plams.Settings()
ss.input.ams.Task = "GeometryOptimization"
ss += engine_s.copy()
slab_jobs = []
for num_layers, slab in zip(num_layers_list, slabs):
millerstring = "".join(str(x) for x in miller)
name = f"{num_layers}layers_{millerstring}"
job = AMSJob(settings=ss.copy(), molecule=slab, name=name)
slab_jobs.append(job)
for job in slab_jobs:
job.run()
import matplotlib.pyplot as plt
import numpy as np
def getresults(job):
main_sys = job.results.get_main_system()
n_atoms = len(main_sys)
energy = job.results.get_energy(unit="hartree")
vecs = main_sys.lattice.vectors
area = np.linalg.norm(np.cross(vecs[0], vecs[1]))
return n_atoms, energy, area
data = sorted(getresults(job) for job in slab_jobs) # sorts on number of atoms
x, y, areas = zip(*data) # x = number of atoms, y = energy
fig, ax = plt.subplots()
ax.plot(x, y, ".")
ax.set_xlim(0, max(x) + 1)
ax.set_ylabel("Energy (hartree)")
ax.set_xlabel("Number of atoms")
ax
ax.figure.savefig("picture4.png")
# We need to fit a straight line through the points and see where it intercepts 0 on the x axis.
# ## Fit a straight line and extrapolate to 0 atoms (pure "surface")
from scipy.stats import linregress
from scm.base import Units
k = 3 # use the last k points
surfacearea = areas[-1] # all should be identical; just use the last number
result = linregress(x[-k:], y[-k:])
lineminx, lineminy = 0, result.intercept
linemaxx, linemaxy = max(x), result.slope * max(x) + result.intercept
fig, ax = plt.subplots()
ax.plot(x, y, ".")
ax.plot([lineminx, linemaxx], [lineminy, linemaxy], "--")
ax.set_xlim(0, max(x) + 1)
ax.set_ylabel("Energy (hartree)")
ax.set_xlabel("Number of atoms")
ax
ax.figure.savefig("picture5.png")
# ## Extract surface energy from y-axis intercept
hartree2J = Units.conversion_factor("hartree", "J")
angstrom2m = Units.conversion_factor("angstrom", "m")
conversion = hartree2J / angstrom2m**2
Esurf = conversion * result.intercept / (2 * surfacearea)
print(f"Intercept = {result.intercept:.6f} hartree")
print(f"Surface energy: {Esurf:.3f} J/m^2")
# ## Final notes on surface energy
# * In the calculation of the surface energy, we never used the energy of the bulk! This is best practice.
#
# * The surface energy is expressed in J/m^2; the area of the unit cell will depend strongly on the optimized the lattice parameters.