In-cell adsorption energies with ReaxFF¶
Compare the standard and in-cell approaches for adsorption energies in ReaxFF.
Build ZnO(10-10) slab supercells of increasing size
Compute E_slab, E_mol, and E_slab+mol widely separated
Quantify E_slab + E_mol - E_slab+mol widely separated
Plot how this difference grows with slab size
In-cell adsorption energies with ReaxFF¶
This example is structured in three parts:
One worked standard adsorption-energy calculation.
One worked in-cell calculation and direct comparison.
Automation over multiple slab sizes to generate a table and graph.
Sign convention used here: adsorption is more stable when the adsorption energy is more negative.
from typing import Dict, List
import matplotlib.pyplot as plt
import pandas as pd
from scm.base import ChemicalSystem
from scm.plams import AMSJob, Settings, view
slab_system = ChemicalSystem("""
System
Atoms
O 1.64000002 -1.90997923 -14.20281662
Zn 1.64000002 0.00000000 -14.20281662
O 0.00000002 0.73002174 -13.25596218
Zn 0.00000002 -2.64000318 -13.25596218
O 0.00000002 -1.90997923 -11.36225329
Zn 0.00000002 0.00000000 -11.36225329
O 1.64000001 0.73002174 -10.41539885
Zn 1.64000001 -2.64000318 -10.41539885
O 1.64000001 -1.90997923 -8.52168997
Zn 1.64000001 0.00000000 -8.52168997
O 0.00000001 0.73002174 -7.57483553
Zn 0.00000001 -2.64000318 -7.57483553
O 0.00000001 -1.90997923 -5.68112665
Zn 0.00000001 0.00000000 -5.68112665
O 1.64000000 0.73002174 -4.73427221
Zn 1.64000000 -2.64000318 -4.73427221
O 1.64000000 -1.90997923 -2.84056332
Zn 1.64000000 0.00000000 -2.84056332
O 0.00000000 0.73002174 -1.89370888
Zn 0.00000000 -2.64000318 -1.89370888
End
Lattice
3.28000000 0.00000000 0.00000000
0.00000000 5.28000000 0.00000000
End
End
""")
def add_water(
system: ChemicalSystem, x: float = 0.0, y: float = 0.0, z: float = 0.0
) -> ChemicalSystem:
ret = system.copy()
ret.add_atom("O", coords=[x + 0.0, y + 0.0, z + 0.59372000])
ret.add_atom("H", coords=[x + 0.0, y + 0.76544, z - 0.00836])
ret.add_atom("H", coords=[x + 0.0, y - 0.76544, z - 0.00836])
return ret
def reaxff_singlepoint_settings() -> Settings:
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.ReaxFF.ForceField = "ZnOH.ff"
s.runscript.nproc = 1
return s
1) One worked example: standard adsorption energy¶
For one slab size, the standard approach uses three calculations:
E_slabE_molE_slab+mol
with
E_ads_standard = E_slab+mol - E_slab - E_mol
With this convention, a negative value means stronger (more stable) adsorption.
example_n = 3
z_ads = 0.0
slab_example = slab_system.make_supercell([example_n, example_n])
mol_example = add_water(ChemicalSystem(), z=z_ads)
slab_and_mol_example = add_water(slab_example, z=z_ads)
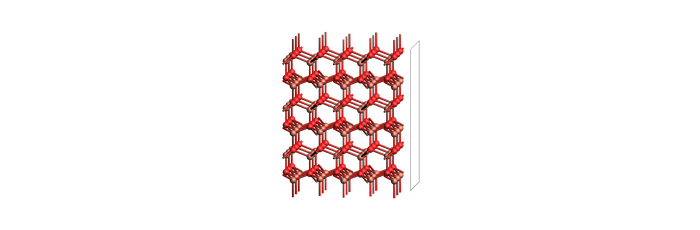
print("Slab")
view(slab_example, width=700, height=230, direction="tilt_x", guess_bonds=True)
Slab
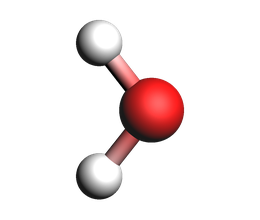
print("Isolated water molecule")
view(mol_example, width=260, height=220, direction="tilt_x", guess_bonds=True)
Isolated water molecule
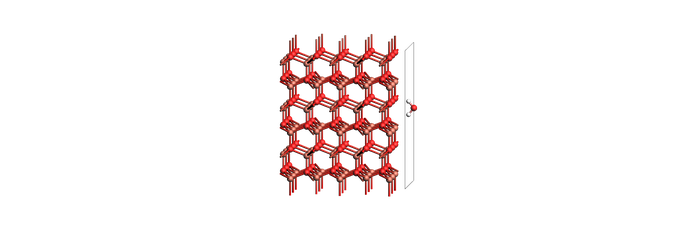
print("Adsorbed slab + molecule")
view(slab_and_mol_example, width=700, height=230, direction="tilt_x", guess_bonds=True)
Adsorbed slab + molecule
s = reaxff_singlepoint_settings()
job_slab = AMSJob(settings=s.copy(), molecule=slab_example, name="example_standard_slab")
job_mol = AMSJob(settings=s.copy(), molecule=mol_example, name="example_standard_mol")
job_ads = AMSJob(
settings=s.copy(), molecule=slab_and_mol_example, name="example_standard_slab_and_mol"
)
job_slab.run()
job_mol.run()
job_ads.run()
E_slab = job_slab.results.get_energy(unit="kcal/mol")
E_mol = job_mol.results.get_energy(unit="kcal/mol")
E_slab_and_mol = job_ads.results.get_energy(unit="kcal/mol")
E_ads_standard = E_slab_and_mol - E_slab - E_mol
df_standard = pd.DataFrame(
[
{"Quantity": "E_slab", "kcal/mol": E_slab},
{"Quantity": "E_mol", "kcal/mol": E_mol},
{"Quantity": "E_slab+mol", "kcal/mol": E_slab_and_mol},
{"Quantity": "E_ads_standard (negative = more stable)", "kcal/mol": E_ads_standard},
]
)
df_standard
[05.03|07:19:38] JOB example_standard_slab STARTED
[05.03|07:19:38] JOB example_standard_slab RUNNING
[05.03|07:19:38] JOB example_standard_slab FINISHED
[05.03|07:19:38] JOB example_standard_slab SUCCESSFUL
[05.03|07:19:38] JOB example_standard_mol STARTED
[05.03|07:19:38] JOB example_standard_mol RUNNING
[05.03|07:19:39] JOB example_standard_mol FINISHED
[05.03|07:19:39] JOB example_standard_mol SUCCESSFUL
[05.03|07:19:39] JOB example_standard_slab_and_mol STARTED
[05.03|07:19:39] JOB example_standard_slab_and_mol RUNNING
[05.03|07:19:39] JOB example_standard_slab_and_mol FINISHED
[05.03|07:19:39] JOB example_standard_slab_and_mol SUCCESSFUL
| Quantity | kcal/mol | |
|---|---|---|
| 0 | E_slab | -15404.569450 |
| 1 | E_mol | -248.178898 |
| 2 | E_slab+mol | -15663.035306 |
| 3 | E_ads_standard (negative = more stable) | -10.286958 |
2) One worked example: in-cell approach¶
For the same slab size, the in-cell approach uses two structures:
E_slab+mol(adsorbed geometry)E_slab+mol_widely_separated
with
E_ads_incell = E_slab+mol - E_slab+mol_widely_separated
Again, with this convention, more negative means more stable adsorption.
z_widely_separated = 30.0
slab_and_mol_widely_example = add_water(slab_example, z=z_widely_separated)
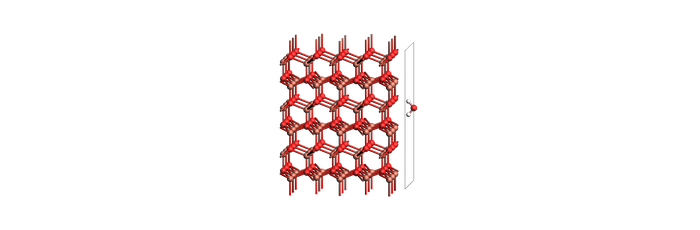
print("Adsorbed slab + molecule (same as above)")
view(slab_and_mol_example, width=700, height=230, direction="tilt_x", guess_bonds=True)
Adsorbed slab + molecule (same as above)
print("Slab + molecule widely separated in the same cell")
view(slab_and_mol_widely_example, width=700, height=230, direction="tilt_x", guess_bonds=True)
Slab + molecule widely separated in the same cell

job_widely = AMSJob(
settings=s.copy(),
molecule=slab_and_mol_widely_example,
name="example_incell_slab_and_mol_widely_separated",
)
job_widely.run()
E_slab_and_mol_widely = job_widely.results.get_energy(unit="kcal/mol")
E_ads_incell = E_slab_and_mol - E_slab_and_mol_widely
difference_incell_minus_standard = E_ads_incell - E_ads_standard
df_compare = pd.DataFrame(
[
{"Approach": "Standard", "E_ads_kcal_mol (negative = more stable)": E_ads_standard},
{"Approach": "In-cell", "E_ads_kcal_mol (negative = more stable)": E_ads_incell},
{
"Approach": "Difference (in-cell - standard)",
"E_ads_kcal_mol (negative = more stable)": difference_incell_minus_standard,
},
]
)
df_compare
[05.03|07:19:41] JOB example_incell_slab_and_mol_widely_separated STARTED
[05.03|07:19:41] JOB example_incell_slab_and_mol_widely_separated RUNNING
[05.03|07:19:41] JOB example_incell_slab_and_mol_widely_separated FINISHED
[05.03|07:19:41] JOB example_incell_slab_and_mol_widely_separated SUCCESSFUL
| Approach | E_ads_kcal_mol (negative = more stable) | |
|---|---|---|
| 0 | Standard | -10.286958 |
| 1 | In-cell | 0.652623 |
| 2 | Difference (in-cell - standard) | 10.939581 |
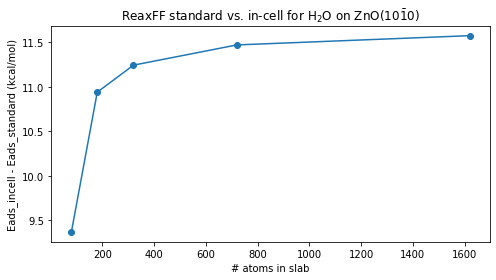
3) Automate over slab sizes (table + graph)¶
Now we repeat this for several supercell sizes and collect all energies in one DataFrame.
supercells: List[int] = [2, 3, 4, 6, 9]
rows = []
for n in supercells:
slab_cs = slab_system.make_supercell([n, n])
mol_cs = add_water(ChemicalSystem(), z=z_ads)
slab_and_mol_cs = add_water(slab_cs, z=z_ads)
slab_and_mol_widely_cs = add_water(slab_cs, z=z_widely_separated)
slab_job = AMSJob(settings=s.copy(), molecule=slab_cs, name=f"auto_slab_{n}")
mol_job = AMSJob(settings=s.copy(), molecule=mol_cs, name=f"auto_mol_{n}")
ads_job = AMSJob(settings=s.copy(), molecule=slab_and_mol_cs, name=f"auto_slab_and_mol_{n}")
widely_job = AMSJob(
settings=s.copy(), molecule=slab_and_mol_widely_cs, name=f"auto_widely_{n}"
)
slab_job.run()
mol_job.run()
ads_job.run()
widely_job.run()
e_slab = slab_job.results.get_energy(unit="kcal/mol")
e_mol = mol_job.results.get_energy(unit="kcal/mol")
e_ads_struct = ads_job.results.get_energy(unit="kcal/mol")
e_widely = widely_job.results.get_energy(unit="kcal/mol")
eads_standard = e_ads_struct - e_slab - e_mol
eads_incell = e_ads_struct - e_widely
rows.append(
{
"supercell_n": n,
"n_atoms_in_slab": len(slab_cs),
"Eslab_kcal_mol": e_slab,
"Emol_kcal_mol": e_mol,
"Eslab_and_mol_kcal_mol": e_ads_struct,
"Eslab_and_mol_widely_separated_kcal_mol": e_widely,
"Eads_standard_kcal_mol (negative=more stable)": eads_standard,
"Eads_incell_kcal_mol (negative=more stable)": eads_incell,
"Eads_incell_minus_standard_kcal_mol": eads_incell - eads_standard,
}
)
df = pd.DataFrame(rows).sort_values("n_atoms_in_slab").reset_index(drop=True)
df
[05.03|07:19:41] JOB auto_slab_2 STARTED
[05.03|07:19:41] JOB auto_slab_2 RUNNING
[05.03|07:19:41] JOB auto_slab_2 FINISHED
[05.03|07:19:41] JOB auto_slab_2 SUCCESSFUL
[05.03|07:19:41] JOB auto_mol_2 STARTED
[05.03|07:19:41] Job auto_mol_2 previously run as example_standard_mol, using old results
[05.03|07:19:41] JOB auto_mol_2 COPIED
[05.03|07:19:41] JOB auto_slab_and_mol_2 STARTED
[05.03|07:19:41] JOB auto_slab_and_mol_2 RUNNING
[05.03|07:19:42] JOB auto_slab_and_mol_2 FINISHED
[05.03|07:19:42] JOB auto_slab_and_mol_2 SUCCESSFUL
[05.03|07:19:42] JOB auto_widely_2 STARTED
... output trimmed ....
[05.03|07:19:43] JOB auto_slab_9 SUCCESSFUL
[05.03|07:19:43] JOB auto_mol_9 STARTED
[05.03|07:19:43] Job auto_mol_9 previously run as example_standard_mol, using old results
[05.03|07:19:43] JOB auto_mol_9 COPIED
[05.03|07:19:43] JOB auto_slab_and_mol_9 STARTED
[05.03|07:19:43] JOB auto_slab_and_mol_9 RUNNING
[05.03|07:19:43] JOB auto_slab_and_mol_9 FINISHED
[05.03|07:19:43] JOB auto_slab_and_mol_9 SUCCESSFUL
[05.03|07:19:43] JOB auto_widely_9 STARTED
[05.03|07:19:43] JOB auto_widely_9 RUNNING
[05.03|07:19:44] JOB auto_widely_9 FINISHED
[05.03|07:19:44] JOB auto_widely_9 SUCCESSFUL
| supercell_n | n_atoms_in_slab | Eslab_kcal_mol | Emol_kcal_mol | Eslab_and_mol_kcal_mol | Eslab_and_mol_widely_separated_kcal_mol | Eads_standard_kcal_mol (negative=more stable) | Eads_incell_kcal_mol (negative=more stable) | Eads_incell_minus_standard_kcal_mol | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 80 | -6846.475323 | -248.178898 | -7104.163642 | -7104.021828 | -9.509421 | -0.141814 | 9.367607 |
| 1 | 3 | 180 | -15404.569450 | -248.178898 | -15663.035306 | -15663.687930 | -10.286958 | 0.652623 | 10.939581 |
| 2 | 4 | 320 | -27385.901246 | -248.178898 | -27644.479149 | -27645.323622 | -10.399005 | 0.844473 | 11.243477 |
| 3 | 6 | 720 | -61618.277844 | -248.178898 | -61876.932838 | -61877.927853 | -10.476095 | 0.995015 | 11.471110 |
| 4 | 9 | 1620 | -138641.125108 | -248.178898 | -138899.815004 | -138900.879160 | -10.510998 | 1.064156 | 11.575154 |
fig, ax = plt.subplots(figsize=(7, 4))
ax.plot(df["n_atoms_in_slab"], df["Eads_incell_minus_standard_kcal_mol"], "o-")
ax.set_xlabel("# atoms in slab")
ax.set_ylabel("Eads_incell - Eads_standard (kcal/mol)")
ax.set_title(r"ReaxFF standard vs. in-cell for H$_2$O on ZnO(10$\bar{1}$0)")
fig.tight_layout()
ax;
See also¶
Python Script¶
#!/usr/bin/env python
# coding: utf-8
# ## In-cell adsorption energies with ReaxFF
#
# This example is structured in three parts:
#
# 1. One worked **standard** adsorption-energy calculation.
# 2. One worked **in-cell** calculation and direct comparison.
# 3. Automation over multiple slab sizes to generate a table and graph.
#
# **Sign convention used here:** adsorption is more stable when the adsorption energy is **more negative**.
#
from typing import Dict, List
import matplotlib.pyplot as plt
import pandas as pd
from scm.base import ChemicalSystem
from scm.plams import AMSJob, Settings, view
slab_system = ChemicalSystem(
"""
System
Atoms
O 1.64000002 -1.90997923 -14.20281662
Zn 1.64000002 0.00000000 -14.20281662
O 0.00000002 0.73002174 -13.25596218
Zn 0.00000002 -2.64000318 -13.25596218
O 0.00000002 -1.90997923 -11.36225329
Zn 0.00000002 0.00000000 -11.36225329
O 1.64000001 0.73002174 -10.41539885
Zn 1.64000001 -2.64000318 -10.41539885
O 1.64000001 -1.90997923 -8.52168997
Zn 1.64000001 0.00000000 -8.52168997
O 0.00000001 0.73002174 -7.57483553
Zn 0.00000001 -2.64000318 -7.57483553
O 0.00000001 -1.90997923 -5.68112665
Zn 0.00000001 0.00000000 -5.68112665
O 1.64000000 0.73002174 -4.73427221
Zn 1.64000000 -2.64000318 -4.73427221
O 1.64000000 -1.90997923 -2.84056332
Zn 1.64000000 0.00000000 -2.84056332
O 0.00000000 0.73002174 -1.89370888
Zn 0.00000000 -2.64000318 -1.89370888
End
Lattice
3.28000000 0.00000000 0.00000000
0.00000000 5.28000000 0.00000000
End
End
"""
)
def add_water(system: ChemicalSystem, x: float = 0.0, y: float = 0.0, z: float = 0.0) -> ChemicalSystem:
ret = system.copy()
ret.add_atom("O", coords=[x + 0.0, y + 0.0, z + 0.59372000])
ret.add_atom("H", coords=[x + 0.0, y + 0.76544, z - 0.00836])
ret.add_atom("H", coords=[x + 0.0, y - 0.76544, z - 0.00836])
return ret
def reaxff_singlepoint_settings() -> Settings:
s = Settings()
s.input.ams.Task = "SinglePoint"
s.input.ReaxFF.ForceField = "ZnOH.ff"
s.runscript.nproc = 1
return s
# ## 1) One worked example: standard adsorption energy
#
# For one slab size, the standard approach uses three calculations:
#
# - `E_slab`
# - `E_mol`
# - `E_slab+mol`
#
# with
#
# `E_ads_standard = E_slab+mol - E_slab - E_mol`
#
# With this convention, a negative value means stronger (more stable) adsorption.
#
example_n = 3
z_ads = 0.0
slab_example = slab_system.make_supercell([example_n, example_n])
mol_example = add_water(ChemicalSystem(), z=z_ads)
slab_and_mol_example = add_water(slab_example, z=z_ads)
print("Slab")
view(slab_example, width=700, height=230, direction="tilt_x", guess_bonds=True, picture_path="picture1.png")
print("Isolated water molecule")
view(mol_example, width=260, height=220, direction="tilt_x", guess_bonds=True, picture_path="picture2.png")
print("Adsorbed slab + molecule")
view(slab_and_mol_example, width=700, height=230, direction="tilt_x", guess_bonds=True, picture_path="picture3.png")
s = reaxff_singlepoint_settings()
job_slab = AMSJob(settings=s.copy(), molecule=slab_example, name="example_standard_slab")
job_mol = AMSJob(settings=s.copy(), molecule=mol_example, name="example_standard_mol")
job_ads = AMSJob(settings=s.copy(), molecule=slab_and_mol_example, name="example_standard_slab_and_mol")
job_slab.run()
job_mol.run()
job_ads.run()
E_slab = job_slab.results.get_energy(unit="kcal/mol")
E_mol = job_mol.results.get_energy(unit="kcal/mol")
E_slab_and_mol = job_ads.results.get_energy(unit="kcal/mol")
E_ads_standard = E_slab_and_mol - E_slab - E_mol
df_standard = pd.DataFrame(
[
{"Quantity": "E_slab", "kcal/mol": E_slab},
{"Quantity": "E_mol", "kcal/mol": E_mol},
{"Quantity": "E_slab+mol", "kcal/mol": E_slab_and_mol},
{"Quantity": "E_ads_standard (negative = more stable)", "kcal/mol": E_ads_standard},
]
)
df_standard
# ## 2) One worked example: in-cell approach
#
# For the same slab size, the in-cell approach uses two structures:
#
# - `E_slab+mol` (adsorbed geometry)
# - `E_slab+mol_widely_separated`
#
# with
#
# `E_ads_incell = E_slab+mol - E_slab+mol_widely_separated`
#
# Again, with this convention, more negative means more stable adsorption.
#
z_widely_separated = 30.0
slab_and_mol_widely_example = add_water(slab_example, z=z_widely_separated)
print("Adsorbed slab + molecule (same as above)")
view(slab_and_mol_example, width=700, height=230, direction="tilt_x", guess_bonds=True, picture_path="picture4.png")
print("Slab + molecule widely separated in the same cell")
view(
slab_and_mol_widely_example,
width=700,
height=230,
direction="tilt_x",
guess_bonds=True,
picture_path="picture5.png",
)
job_widely = AMSJob(
settings=s.copy(),
molecule=slab_and_mol_widely_example,
name="example_incell_slab_and_mol_widely_separated",
)
job_widely.run()
E_slab_and_mol_widely = job_widely.results.get_energy(unit="kcal/mol")
E_ads_incell = E_slab_and_mol - E_slab_and_mol_widely
difference_incell_minus_standard = E_ads_incell - E_ads_standard
df_compare = pd.DataFrame(
[
{"Approach": "Standard", "E_ads_kcal_mol (negative = more stable)": E_ads_standard},
{"Approach": "In-cell", "E_ads_kcal_mol (negative = more stable)": E_ads_incell},
{
"Approach": "Difference (in-cell - standard)",
"E_ads_kcal_mol (negative = more stable)": difference_incell_minus_standard,
},
]
)
df_compare
# ## 3) Automate over slab sizes (table + graph)
#
# Now we repeat this for several supercell sizes and collect all energies in one DataFrame.
#
supercells: List[int] = [2, 3, 4, 6, 9]
rows = []
for n in supercells:
slab_cs = slab_system.make_supercell([n, n])
mol_cs = add_water(ChemicalSystem(), z=z_ads)
slab_and_mol_cs = add_water(slab_cs, z=z_ads)
slab_and_mol_widely_cs = add_water(slab_cs, z=z_widely_separated)
slab_job = AMSJob(settings=s.copy(), molecule=slab_cs, name=f"auto_slab_{n}")
mol_job = AMSJob(settings=s.copy(), molecule=mol_cs, name=f"auto_mol_{n}")
ads_job = AMSJob(settings=s.copy(), molecule=slab_and_mol_cs, name=f"auto_slab_and_mol_{n}")
widely_job = AMSJob(settings=s.copy(), molecule=slab_and_mol_widely_cs, name=f"auto_widely_{n}")
slab_job.run()
mol_job.run()
ads_job.run()
widely_job.run()
e_slab = slab_job.results.get_energy(unit="kcal/mol")
e_mol = mol_job.results.get_energy(unit="kcal/mol")
e_ads_struct = ads_job.results.get_energy(unit="kcal/mol")
e_widely = widely_job.results.get_energy(unit="kcal/mol")
eads_standard = e_ads_struct - e_slab - e_mol
eads_incell = e_ads_struct - e_widely
rows.append(
{
"supercell_n": n,
"n_atoms_in_slab": len(slab_cs),
"Eslab_kcal_mol": e_slab,
"Emol_kcal_mol": e_mol,
"Eslab_and_mol_kcal_mol": e_ads_struct,
"Eslab_and_mol_widely_separated_kcal_mol": e_widely,
"Eads_standard_kcal_mol (negative=more stable)": eads_standard,
"Eads_incell_kcal_mol (negative=more stable)": eads_incell,
"Eads_incell_minus_standard_kcal_mol": eads_incell - eads_standard,
}
)
df = pd.DataFrame(rows).sort_values("n_atoms_in_slab").reset_index(drop=True)
df
fig, ax = plt.subplots(figsize=(7, 4))
ax.plot(df["n_atoms_in_slab"], df["Eads_incell_minus_standard_kcal_mol"], "o-")
ax.set_xlabel("# atoms in slab")
ax.set_ylabel("Eads_incell - Eads_standard (kcal/mol)")
ax.set_title(r"ReaxFF standard vs. in-cell for H$_2$O on ZnO(10$\bar{1}$0)")
fig.tight_layout()
ax
ax.figure.savefig("picture6.png")