Periodic Energy Decomposition Analysis - PEDA¶
This tutorial will teach you how to perform a Periodic Energy Decomposition Analysis (PEDA) for periodic systems with BAND.
Set up the system - CO@MgO(100) (√2×√2)-R45°¶
Start AMSinput in a clean directory and switch to Band:
 →
→ 
Mg 0.59538358 0.59538226 0.20750000
Mg 2.08384335 -0.89307752 -1.89750000
Mg -2.38153597 0.59538226 0.20750000
Mg -0.89307620 -0.89307752 -1.89750000
O 2.08384335 -0.89307752 0.20750000
O -0.89307620 -0.89307752 0.20750000
O 0.59538358 0.59538226 -1.89750000
O -2.38153597 0.59538226 -1.89750000
O 0.59538553 0.59539200 3.95250000
C 0.59538493 0.59538903 2.80750000
VEC1 2.97691955 -2.97691955 0.00000000
VEC2 2.97691955 2.97691955 0.00000000
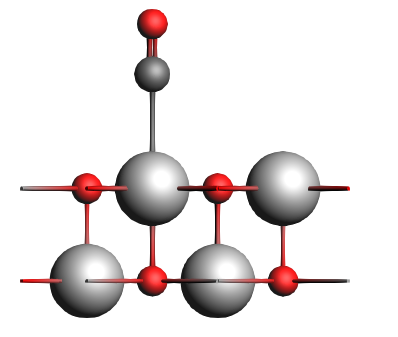
Set up the PEDA calculation¶
To run the PEDA for the adsorption of CO on MgO, you have to define the fragments. Therefore switch to Regions menu.
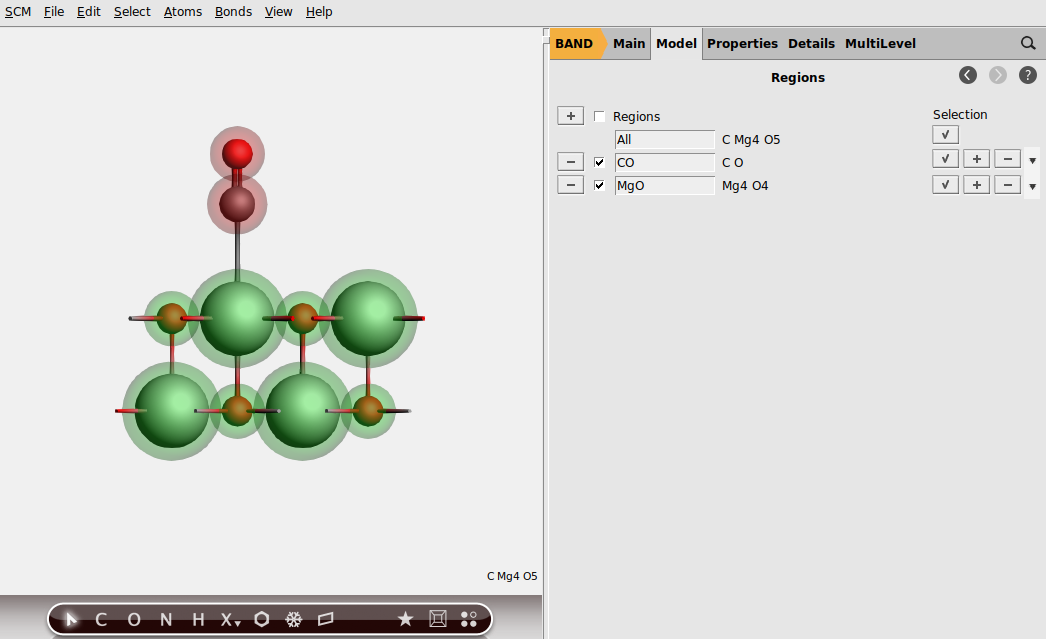
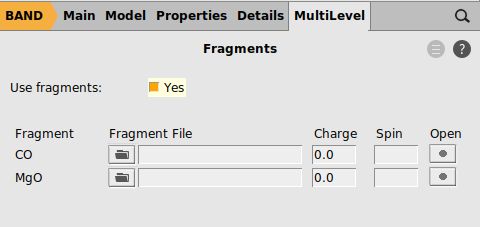
We now enable the PEDA calculation:
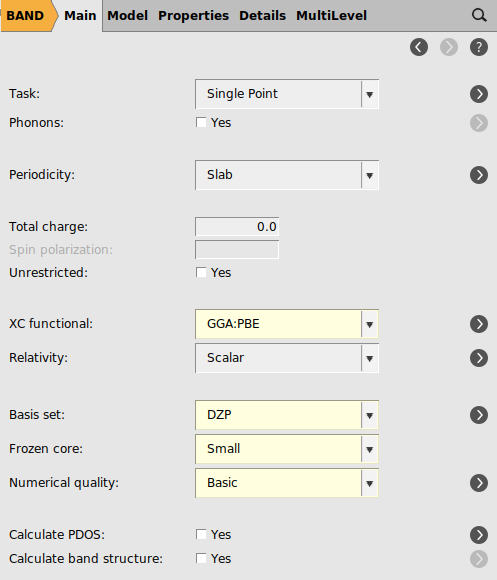
Go back to the main panel set some calculation options for BAND:
Run the calculation check the results¶
Now you can save and run the calculation:
After the calculations of the fragments and the PEDA finished you can look for the PEDA results. Open the “Output” using the SCM dropdown menu:
You can jump to the ‘PEDA Energy Terms’ via the corresponding button in the ‘Properties’ drop-down menu.
Reference results:
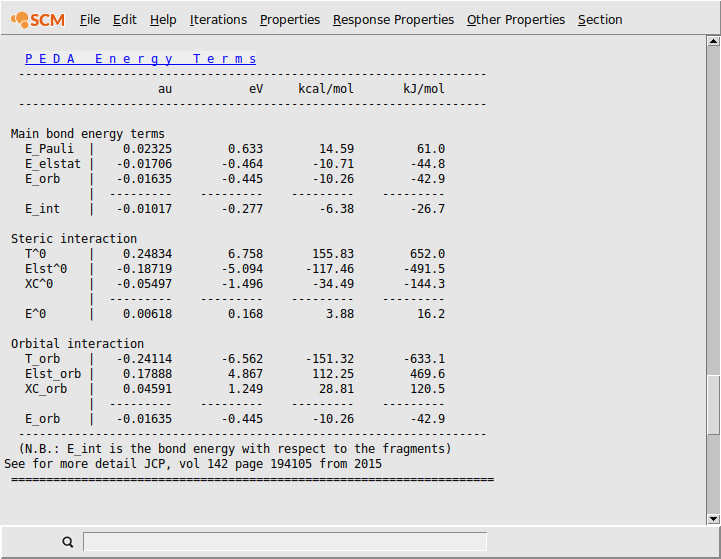
In addition to these energy terms the summed preparation energies of the fragments and the (negative) bond dissociation energy are usually given. Therefore you have to calculate the energy difference between the electronically and structurally relaxed fragments (which can be accessed by a geometry optimization of the separated fragments) and the promoted fragments (which are already calculated and used for the PEDA). Adding this energy difference, which is equal to the preparation energy, to the interaction energy will give you the negative bond dissociation energy.
Plot the deformation density with respect to the fragments¶
Fire up amsview
and select add -> Isosurface: With Phase. At the bottom you can now open the selector, and this shows an extra item “Fragment related densities”. Select “Deformation density w.r.t. sum-of-fragments”. You should see
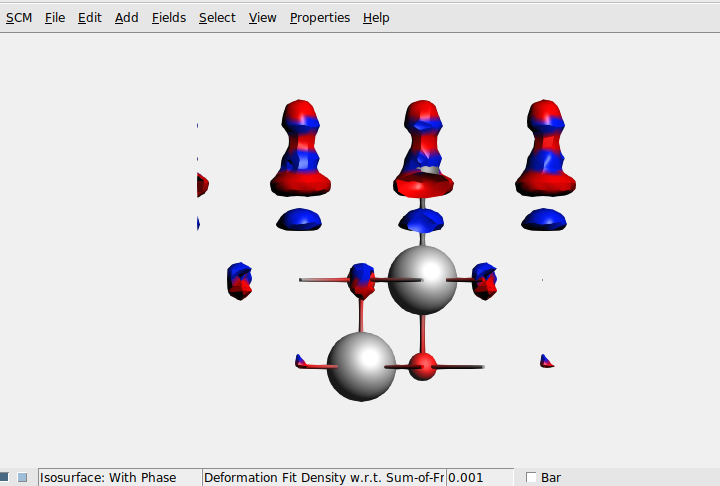
Make sure that you select View→ View Direction→ Along Y-axis, change the isovalue from 0.03 to 0.001, and deselect View→ Periodic→ Show Lattice Vectors. As you can see, more is happening on the CO than on the MgO surface. One could improve the picture by using Fields→ Grid→ Medium, for example.
You can also visualize the same, but obtained with via the fitted density. This is faster, but is an approximation. In this case they look pretty much the same.