QTAIM (Bader), localized orbitals and conceptual DFT¶
This tutorial will show you how to perform a QTAIM analysis, compute localized orbitals and conceptual DFT descriptors using ADF.
QTAIM analysis of an Adenine–Thymine base pair¶
 panel active.
panel active.Next we need the structure of the Adenine–Thymine (AT) base pair:
 and select DNA → AT
and select DNA → ATYou should now see the AT base pair in the molecule drawing area.
Not all QTAIM options can be used in combination with relativistic effects, so in this Tutorial we disable them:
Now we want to activate the QTAIM analysis to find the critical points and bond paths. To find where this option is located, search for it:
 (cmd/ctrl-F)
(cmd/ctrl-F)AMSinput will switch to the panel that displays the option you are looking for (to calculate the AIM critical points and paths). The matching input options will be marked with blue text. Note that the input option search will restrict the search to panels that belong to the current method (ADF, BAND, DFTB, …)
Now we are ready to run the calculation.
A dialog will pop up in which you must specify a filename to use for your job, for example ‘AT’:
After hitting the save button the calculation will start. You will get a new AMSjobs window that allows you to manage your jobs and keep track of their state (for example, queued or running). For a running job, there will be two lines showing the progress. To get more information you can show the logfile in a new window:
Depending on your computer, the calculation should be ready after a few minutes at most.
Once the calculation is finished, use AMSview to visualize the results. To visualize the critical points and bond paths:
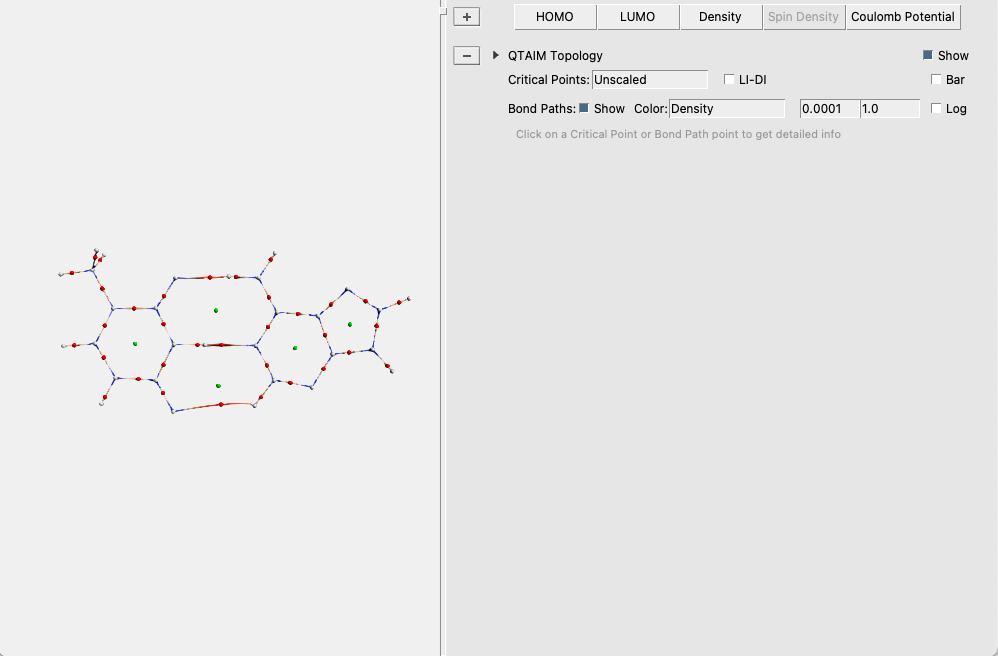
The critical points and bond paths are shown (the molecule balls and sticks representation is hidden). The different types of critical points (atom CP, bond CP, ring CP and cage CP) are indicated by different colors:
atom CP: white
bond CP: red
ring CP: green
The bond paths are colored by density, by default. You can get extra information about a CP or a point along the bond path by clicking on it.
To inspect the basin partitioning itself:
We will now add a plane showing the electronic density:
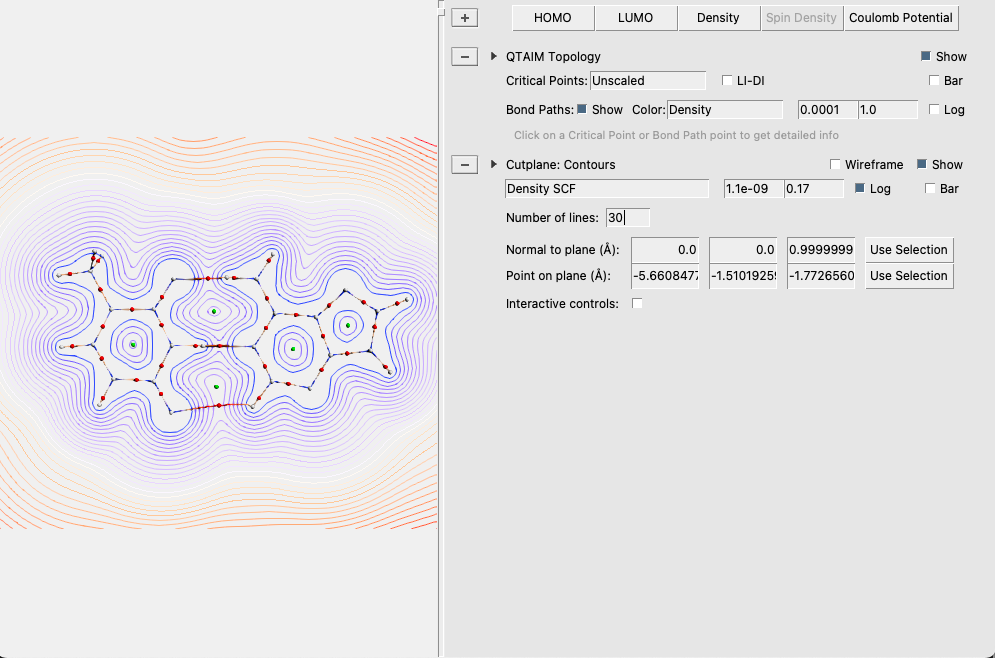
This concludes the AT QTAIM tutorial. To close all its windows:
Benzene QTAIM charge analysis and NBOs¶
When the calculation is done (it should run very fast), we use AMSview to examine the QTAIM charges and compare them with Mulliken charges:
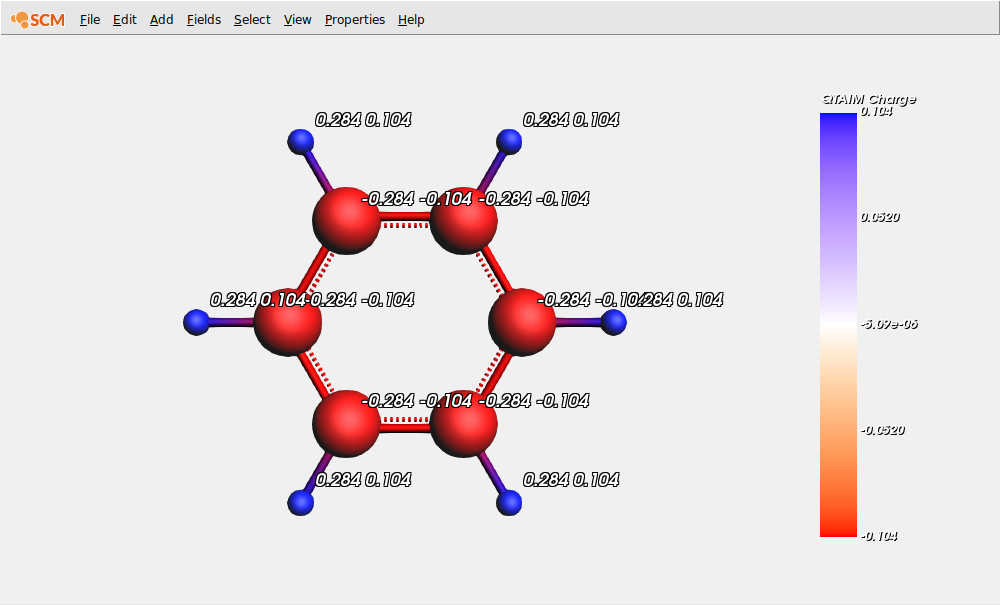
Note
This part requires you to have a license for the NBO6 program. Starting in July 2026, all new AMS2026 and later license files include a license for NBO6. If your AMS2026 or later license file does not include NBO6, please request a new license by contacting license@scm.com.
Next we inspect the NBOs and Boys-Foster localized orbitals. To remove the charge display we close and open AMSview, but you could also have used the View menu to remove them by hand:
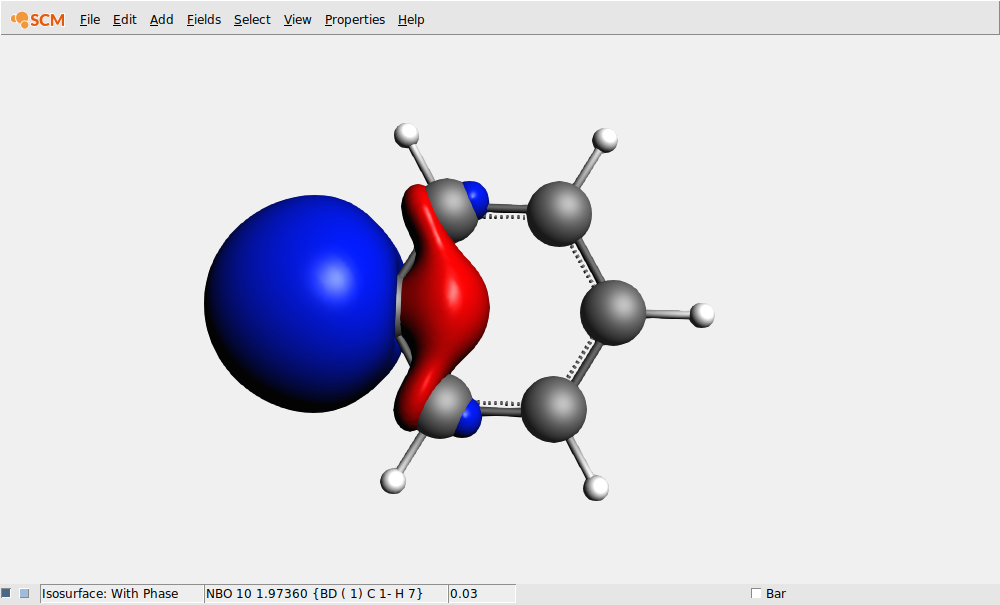
You can also visualize the NLMOs or the Boys-Foster localized orbitals (which are just called Localized Orbitals in the fields menu.
Rationalizing a typical SN2 reaction using condensed Conceptual DFT descriptors¶
The chemical reactivity of reactants or key intermediates can be analyzed using condensed (over QTAIM basins) Conceptual DFT descriptors such as Fukui functions or Dual Descriptor. We strongly suggest the use of the Dual Descriptor, which gives at one glance a more complete description of reactivity behaviors. All the following calculations are based on frontier molecular orbitals (FMOs) using Koopmans approximation, which presents advantages (fast calculations) and drawbacks (in particular if FMOs are degenerated or quasi-degenerated).
An alternative way, based on finite difference linear (FDL) approximation, is available in ADF: Fukui Functions and Dual Descriptor. The FDL approximation offers a more rigorous approach, but it requires three calculations (systems with N electrons (reference), N+δ electrons and N-δ electrons (0<δ<=1)) and shows other drawbacks. For instance, adding one electron to the reference system may lead to unconverged SCF procedure, or the corresponding spin states might be unobvious. Besides, some ambiguity remains about which atomic basins (relaxed or unrelaxed) should be used when adding or removing electrons.

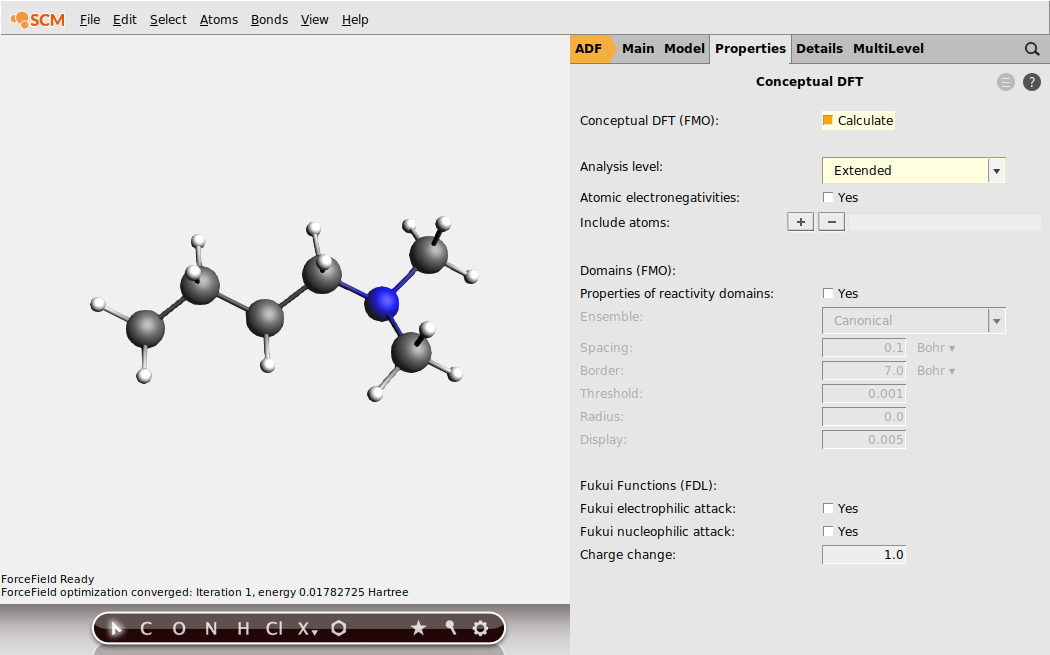
At the end of the optimization process, all the QTAIM properties will be calculated.
Show the condensed (over QTAIM atomic basins) ‘Fukui Fminus function’ indices that characterize the nucleophilicity of atomic sites:
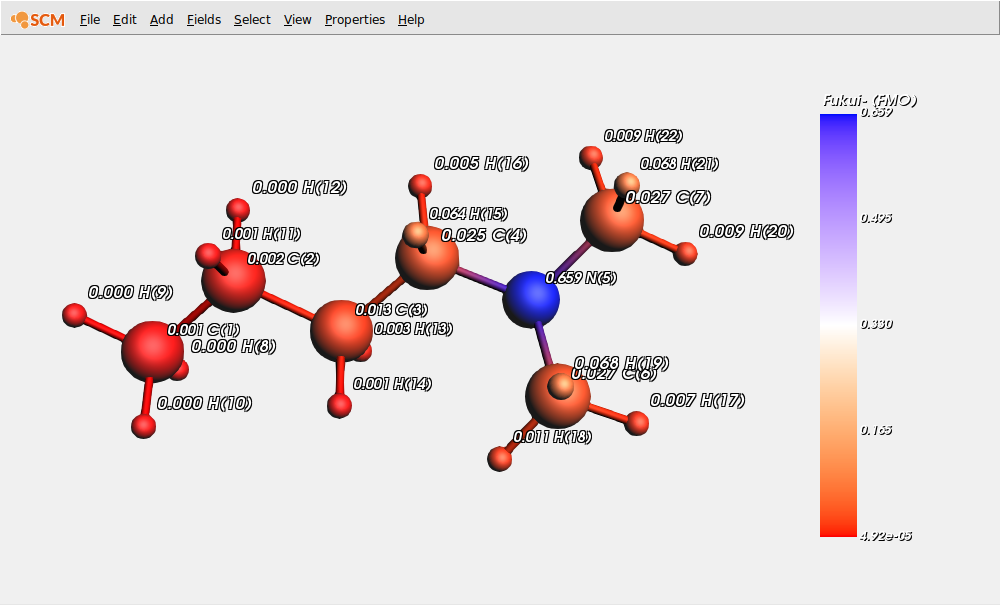
On this picture, we clearly see that the nitrogen site is the most nucleophilic one. To obtain a more complete picture at one glance, we can visualize the condensed values of the dual descriptor (DD) that corresponds, using the Koopmans’ theorem, to the difference between FMOs electron densities.
To this end, first hide the previous values and display the condensed DD values:
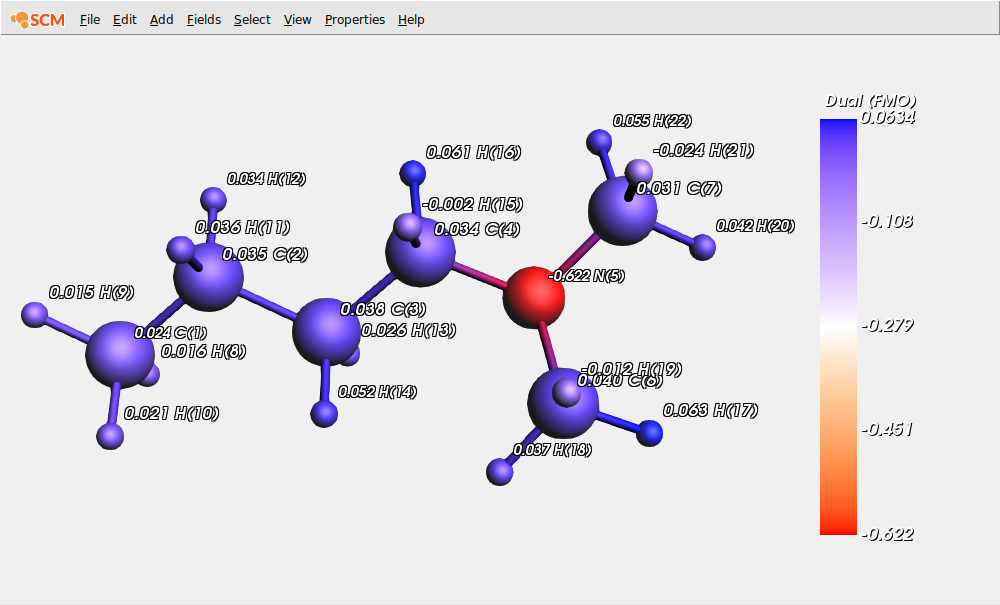
Positive values correspond to atomic sites where electrophilicity is predominant, while negative values correspond to atomic sites where nucleophilicity is predominant (again, the nitrogen atom is highly nucleophilic).
In a new input window, now make the benzyl chloride (electrophile):
C -0.70294970 0.03823073 0.00000000
C -0.02771734 -1.20050280 0.00000000
C 1.37040750 -1.24326069 0.00000000
C 2.10941268 -0.05859271 -0.00000000
C 1.45241936 1.17312771 -0.00000000
C 0.05527963 1.22223527 -0.00000000
C -2.21056076 0.15917615 -0.00000000
Cl -2.96962094 0.22007043 1.61845248
H -0.56397603 -2.13845972 0.00000000
H 1.88164983 -2.19732981 0.00000000
H 3.19110656 -0.09523365 -0.00000000
H 2.02573037 2.09116490 -0.00000000
H -0.43823632 2.18658642 -0.00000000
H -2.49816320 1.08415158 -0.54318756
H -2.64194499 -0.70811986 -0.54318753
At the end of the optimization process, all the QTAIM properties will be calculated.
Show the condensed (over QTAIM atomic basins) ‘Fukui+ (FMO) function’ values that characterize the electrophilicity of atomic sites:
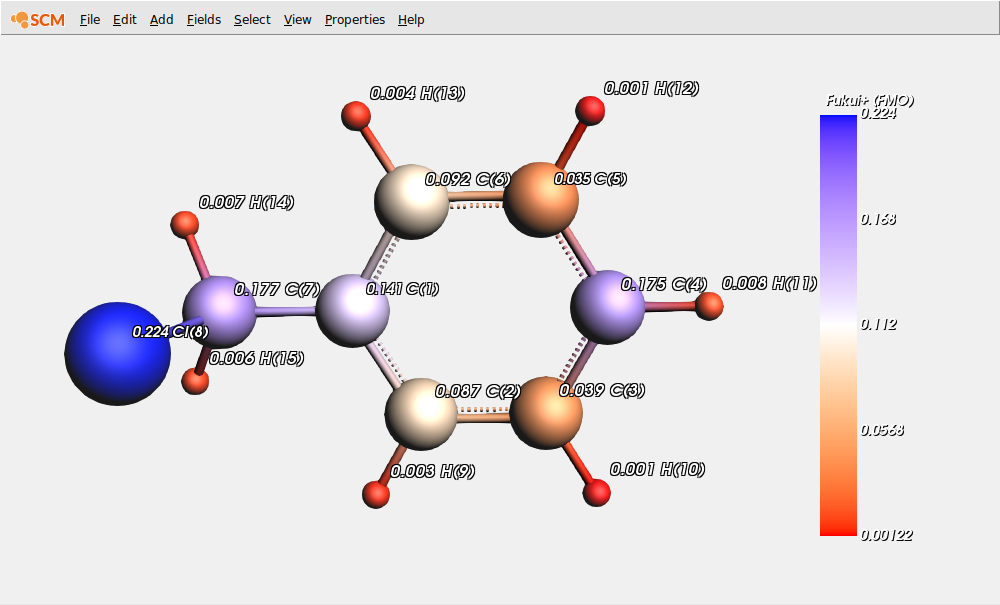
On this picture, two carbon sites (C(4) and C(7)) have similar Fukui+ indices. Moreover, chlorine has a strong electrophilic character due to the existence of a sigma hole in the outer part of its valence shell along the C-Cl bond. Therefore, it is difficult to unambiguously determine the reactivity of this molecule by the sole QTAIM condensed Fukui+ values. In that case, the dual descriptor is quite useful, providing a balanced picture, since it allows evaluating the predominant reactivity behavior at each atomic site.
To this end, first hide the previous values and display the condensed DD values:
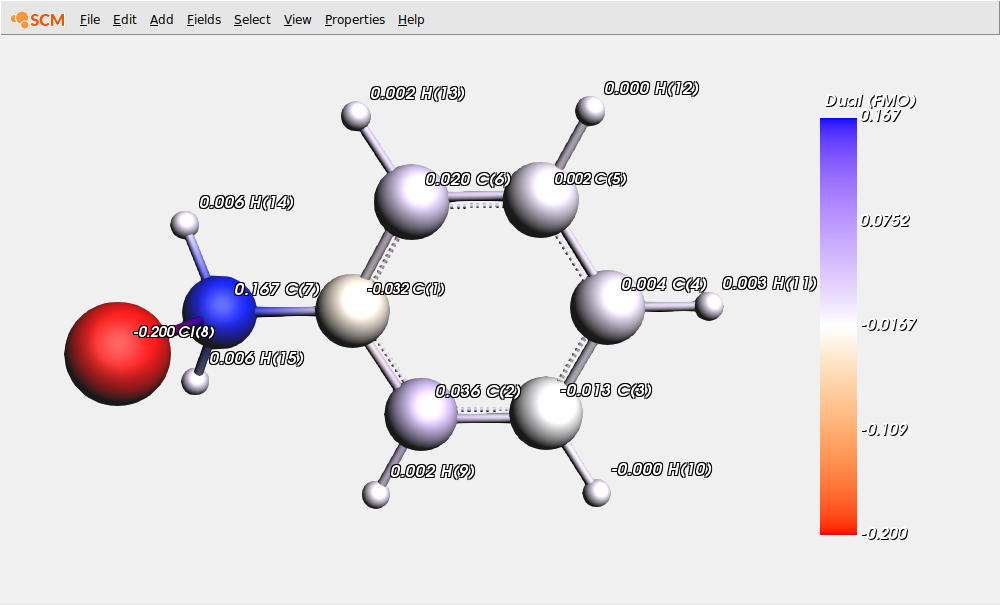
As already mentioned, positive values correspond to atomic sites where electrophilicity is predominant, while negative values correspond to atomic sites where nucleophilicity is predominant.
In this picture we clearly see, as expected from chemical intuition, that C(7) is highly electrophilic (compared to the other carbon atoms). This site will thus undergo a nucleophilic attack during the SN2 reaction with the N,N-dimethylbutylamine molecule, leading to the formation of a quaternary ammonium salt.
Besides, we can also observe that the chlorine atom is predominantly a nucleophilic site (due to its lone pairs) despite the presence of an electrophilic sigma hole.