Restart the DOS or band structure with BAND¶
It is quite likely that you did a calculation, but forgot to request the DOS or BandStructure. Or maybe you did but want to use different settings.
Fortunately it is possible to calculate these directly from the previous calculation.
With the restart of the DOS it is possible to calculate the DOS with a finer k-space sampling. In this example we first show an example where the DOS does not match the band structure. This missing DOS problem can be solved by simply using a better k-grid. However, it can also be solved by doing a restart, using only a better k-grid for the DOS calculation.
Step 1: Load the geometry¶
The have the geometry stored as an xyz file
Mo3WSeS7.xyzLet us load it into AMSinput.
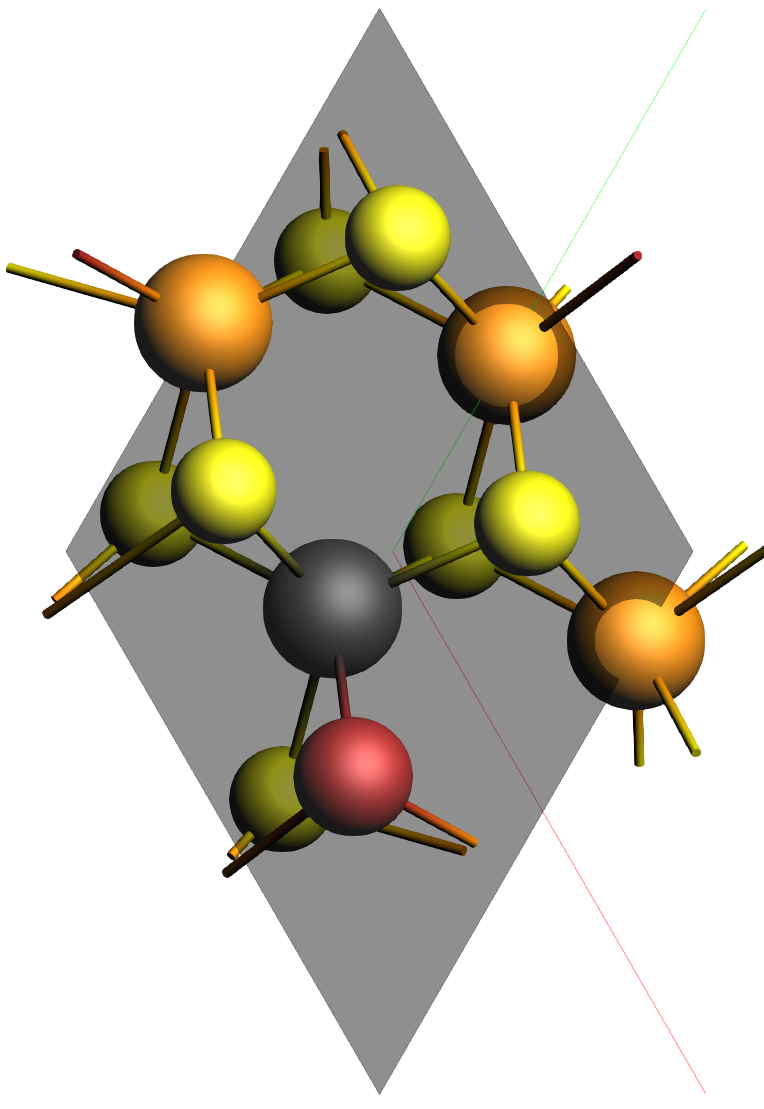
Mo3WSeS7.xyz and click OKThe structure is a MoS2 slab with some substitutions
Step 2: Calculation¶
With the system loaded it is time to setup the calculation
Yes next to Calculate PDOS and Calculate band structure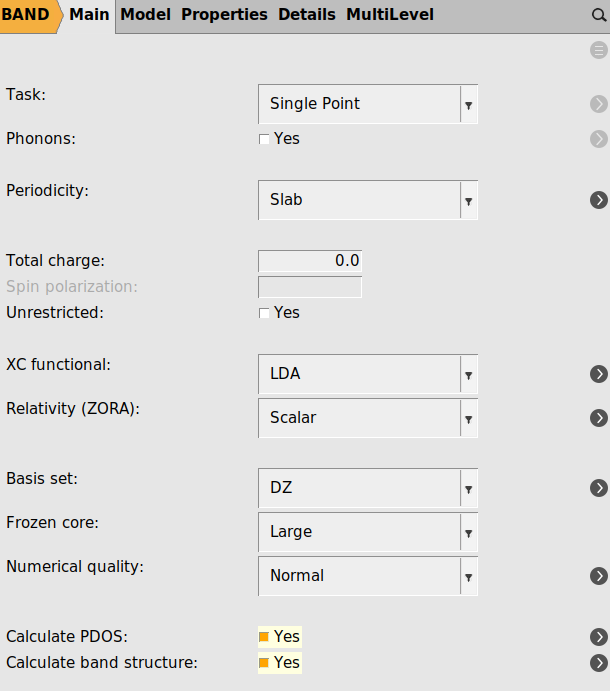
test_k=normal.amsNow run the calculation with File → Run. This calculation may take a few minutes.
Step 3: Inspecting the results¶
After the calculation is complete, open the results with SCM → Band Structure. To highlight the missing DOS problem change the y-range to -8 to -4 eV
Now we can see the problem: there clearly is a band between -5.6 and -5.2 eV, but the DOS is zero in this range.
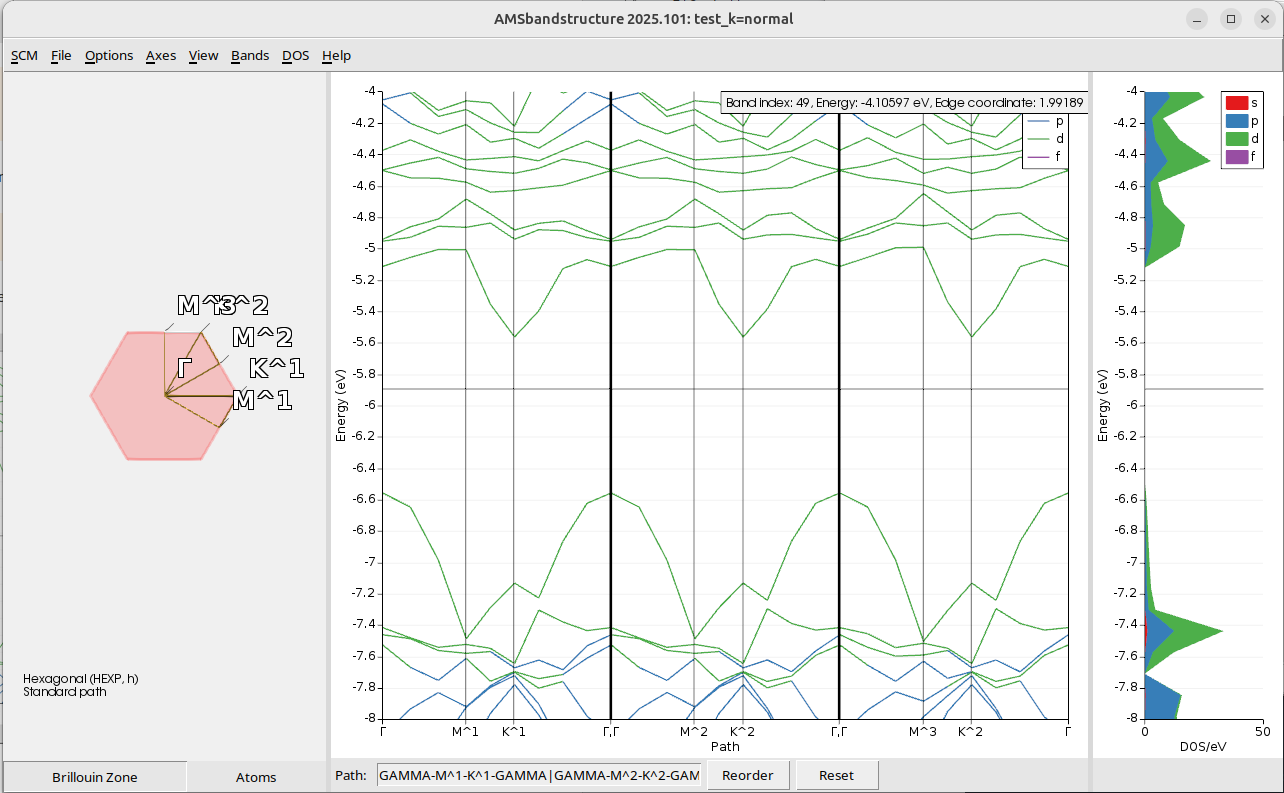
Step 4: The simple solution: more k-points¶
The simple solution is to do the whole calculation with a better k-grid.
test_k=good.amsRun the calculation with File → Run, and this calculation will take a bit longer than before.
and now there is no longer DOS missing in the -5.6 and -5.2 energy range.
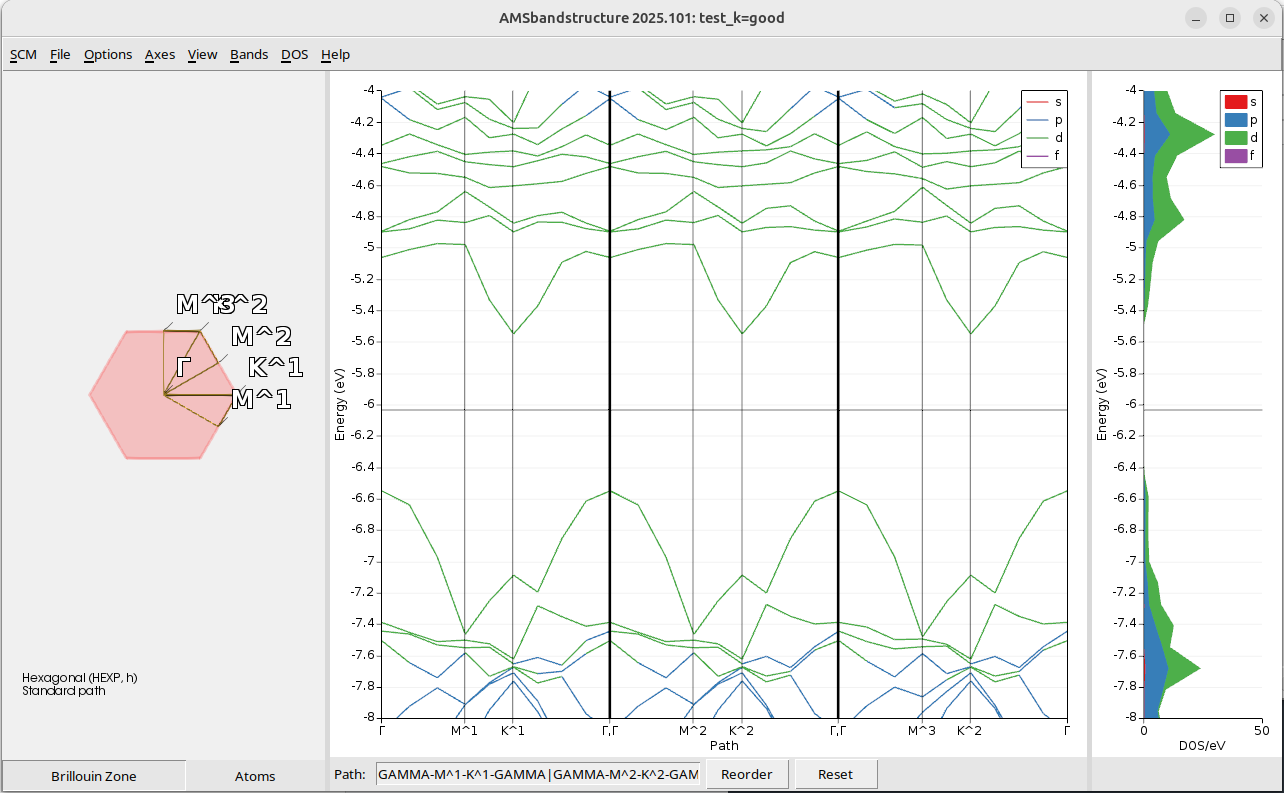
Step 5: Restarting the DOS with more k-points¶
test_k=good.amstest_k=normal.results/band.rkf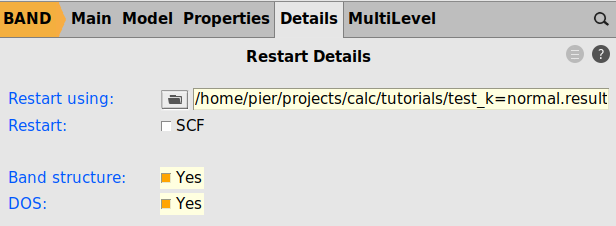
test_k=normal_krestart=goodThe band structure and DOS look very similar to what we had with a good k-grid for the whole calculation (see Step 4), but obtained in a faster way, avoiding the need to do a full SCF with the good k-grid. Note that there is no longer missing dos between -5.6 and -5.2 eV.
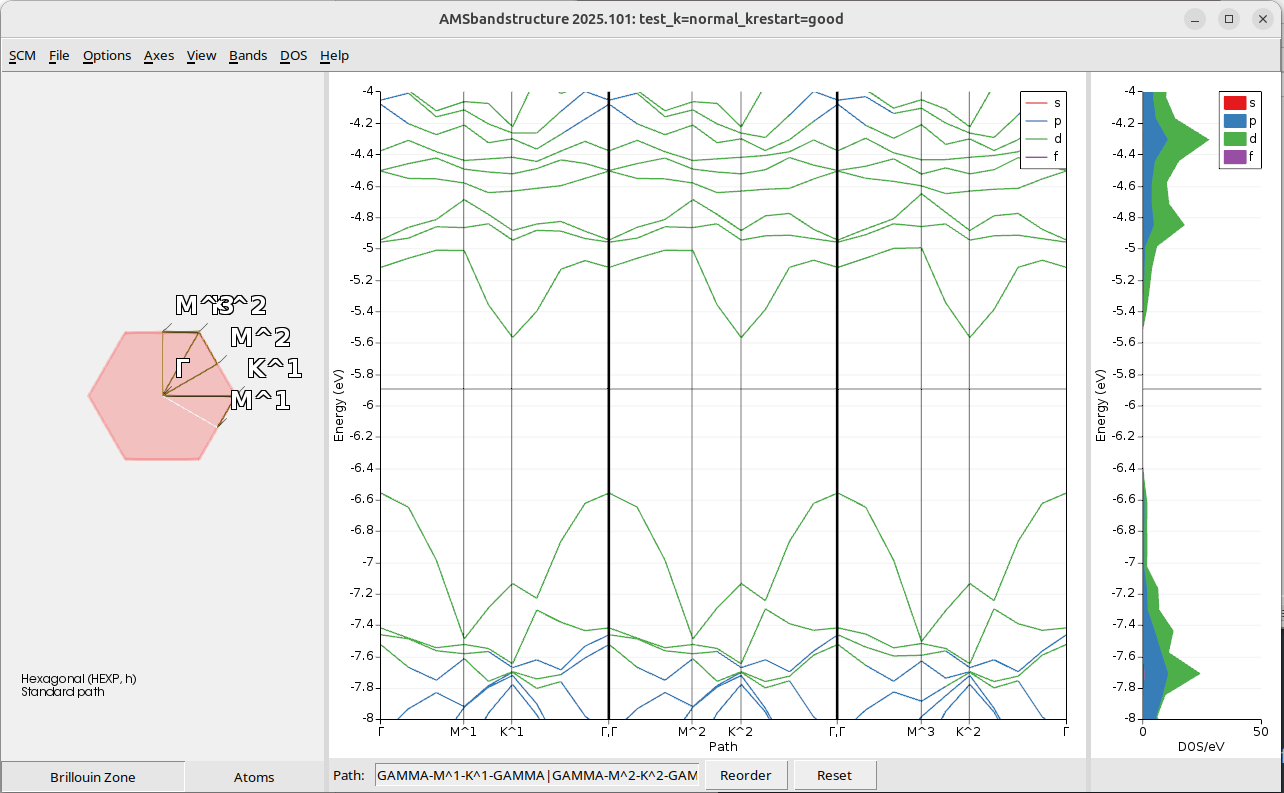
Step 6: Refining the plot¶
In the final step we use more energy points for the dos grid and finer sampling of the band structure
test_k=normal_krestart=good.ams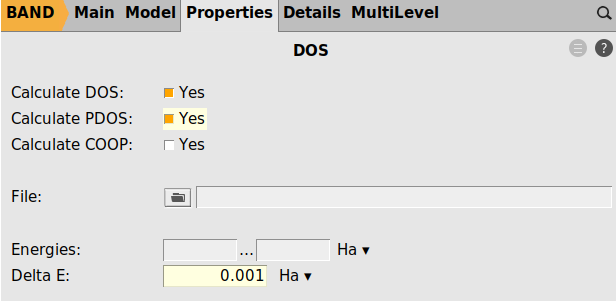
test_k=normal.krestart=good_refined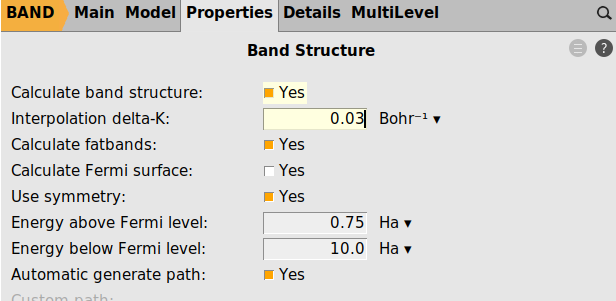
The refined plot is
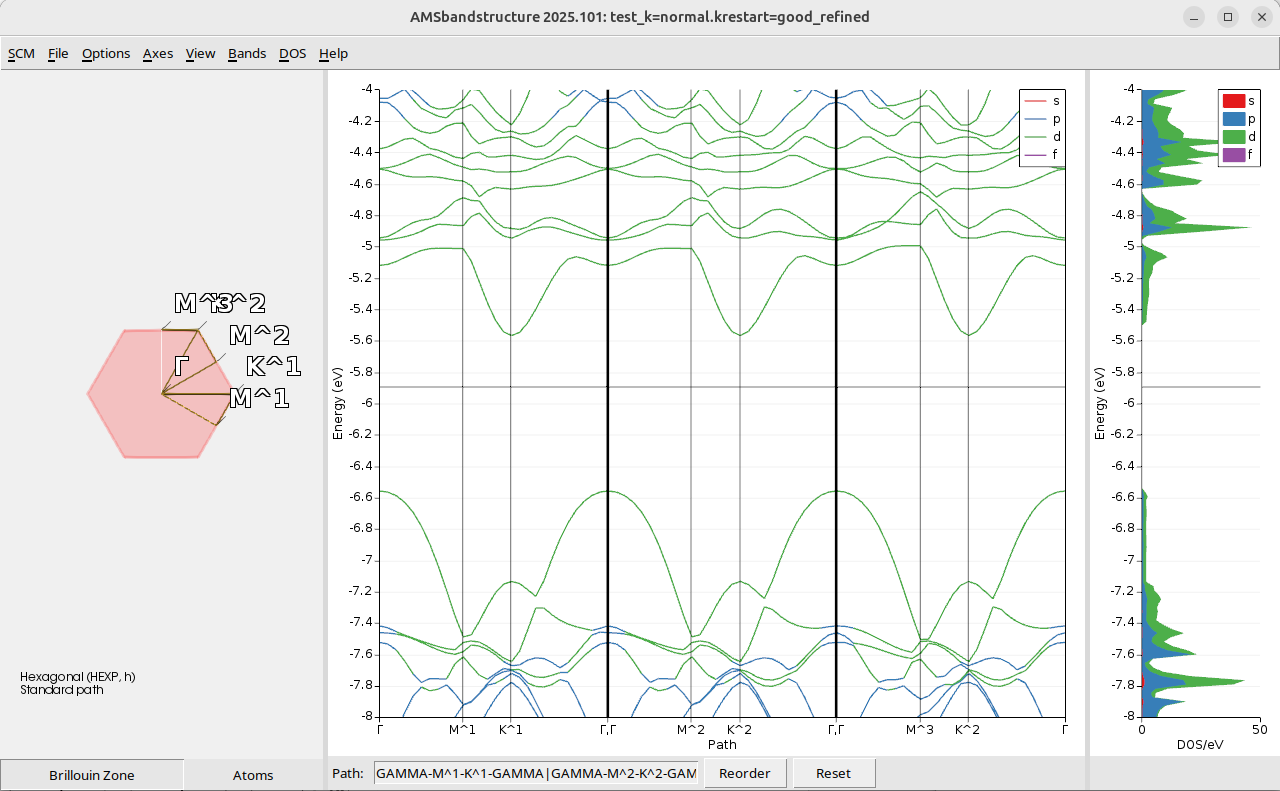
The reference result obtained by simply doing a full SCF with good k-grid and refined DOS DeltaE and BandStructure delta-K is
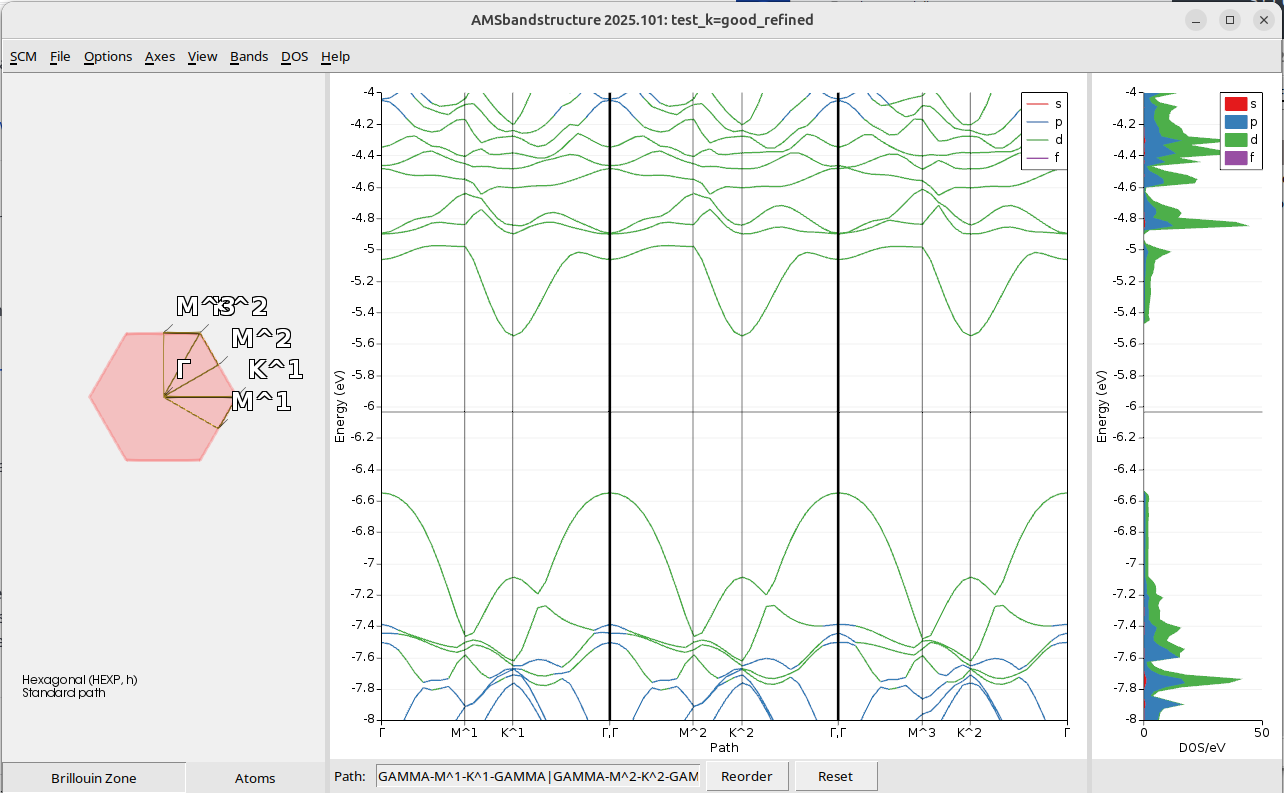
and the results look very similar, so you do not need to perform this calculation.