GW: Ionization Potential and Electron Affinity¶
Ionization potential (IP) and electron affinity (EA) of molecules can be predicted with reasonable accuracy using the G0W0 method [1] [2], especially if as starting point a (range separated) hybrid functional is used. Note that functionals that use XCfun (like LCY-BP86 and CAMY-B3LYP) can not be used as starting point.
One can also do eigenvalue-only self-consistent GW (evGW) with ADF, which is typically a factor 5 to 10 more expensive than G0W0, but which has the advantage that it is less starting-point dependent. Starting from AMS2022 one can also do quasiparticle self-consistent GW (qsGW) with ADF, which is more expensive than evGW, but the results of a qsGW are starting-point independent, and is usually the most accurate one.
Next in AMS2022 it also possible to go beyond the GW approximation for the self-energy and take into account the screened interaction to second order, with an even more expensive method called G3W2. See the ADF manual on GW for more details.
In AMS2023.102 a bug was fixed regarding GW. An incorrect Fermi energy was used, which also affected BSE.
In this tutorial we will show how to compute the IP and EA of Azulene with ADF using G0W0@PBE0.
Warning
The implemented GW algorithm is very sensitive to numerical noise and on the numerical (MBPT) settings. Using the same numerical settings, quasiparticle energies from partially self-consistent GW calculations performed on different hardware can differ by a few meV. See also the ADF manual on GW and ADF manual on MBPT settings.
Set up and run the calculation¶
Let’s start by importing the structure of the Azulene molecule from our molecules database:
 search for
search for Azulene and select C10H8: Azulene
Let’s now set up the calculation details in the main panel:
Typically for EA one may need a larger basis set than TZ2P, see the section Recommendations for GW calculations in the ADF manual. For precision reasons (at least) a good numerical quality is recommended.
To enable the G0W0 calculation:
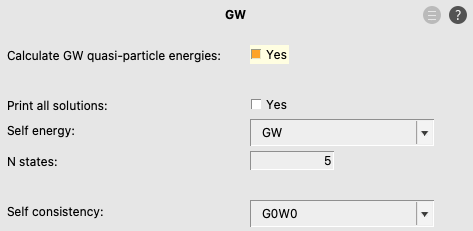
Use Self consistency → EVGW for evGW, and use Self consistency → QSGW for qsGW. Use Self energy → G3W2 for G3W2, typically used in combination with qsGW. In case of (any variant of) GW, in order to avoid numerical issues, the dependency key is set in ADF (and the RIHartreeFock%DependencyThreshold is increased), see the Recommendations for GW calculations in the ADF manual. One can change these dependency values using the panel bar Details → Accuracy.
We are now ready to run the calculation:
Results¶
The results of the calculation are written both to the text output file and to the binary adf.rkf file.
To see the G0W0 results in the output file:
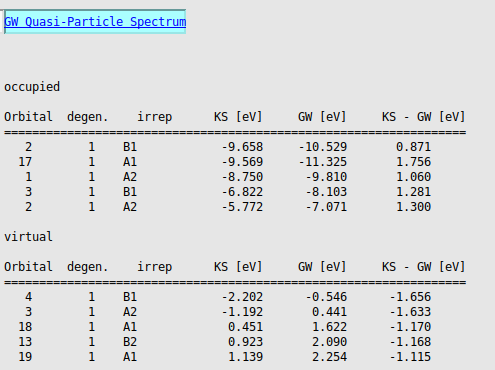
The ionization potential (IP) equals the negative of the G0W0 quasi-particle HOMO energy:
The electron affinity (EA) equals the negative of the G0W0 quasi-particle LUMO energy:
These values are in good agreement with the CCSD(T) results from [1] (IP=7.55 [eV] and EA=0.54 [eV]) and with experimental results cited in [1] (IP=7.42-7.44 [eV] and EA=0.69-0.80 [eV]).