QM/FQ coupled to MD: UV/Vis absorption in aqueous solution¶
In this tutorial, we will simulate the UV-vis absorption spectrum of 2-Methyloxirane in water using the QM/FQ method. There are two major steps:
Molecular dynamics (MD) simulations to sample the solute-solvent configurational space
QM/FQ calculations on MD snapshots to obtain the UV/vis absorption spectrum.
Both the correct sampling of the configurational space and the subsequent QM calculations upon the solvated molecules are crucial to the quality of the end results.
Key references:
Giovanni et al, “Molecular spectroscopy of aqueous solutions: a theoretical perspective” Chem. Soc. Rev. 49 (2020) 5664.
Giovannini et al, “Theory and algorithms for chiroptical properties and spectroscopies of aqueous systems” Phys. Chem. Chem. Phys. 22 (2020) 22864,
Cappelli, “Integrated QM/polarizable MM/continuum approaches to model chiroptical properties of strongly interacting solute-solvent systems” Int. J. Quantum Chem. 116 (2016) 1532.
The tutorial is divided into two stages: first, we set/run the MD, and then we set/run the QM/FQ calculations. Furthermore, we have two routes:
Route # 1: Written for standard AMS users. Both MD (in AMS) and QM/FQ simulations are explained.
Route # 2: Written for those who already run the MD simulation either in AMS suite or in another package like GROMACS, AMBER, etc, as long as they have any snapshot. This route starts in Step 4, item b, where the QM/FQ simulations are outlined.
Route # 1 (AMS users)
For this simulation, we will use the Force Field engine through the AMS driver program.
See also
For new users, see the the Getting started Tutorial and Building Molecules Tutorial.
Step 1: Building the system¶
 →
→ 
We will build a periodic box with one 2-methyloxirane molecule solvated by water.
1.0 g/cm³.30, 30, 30 Å.CC1CO11 to add a new row
to add a new rowO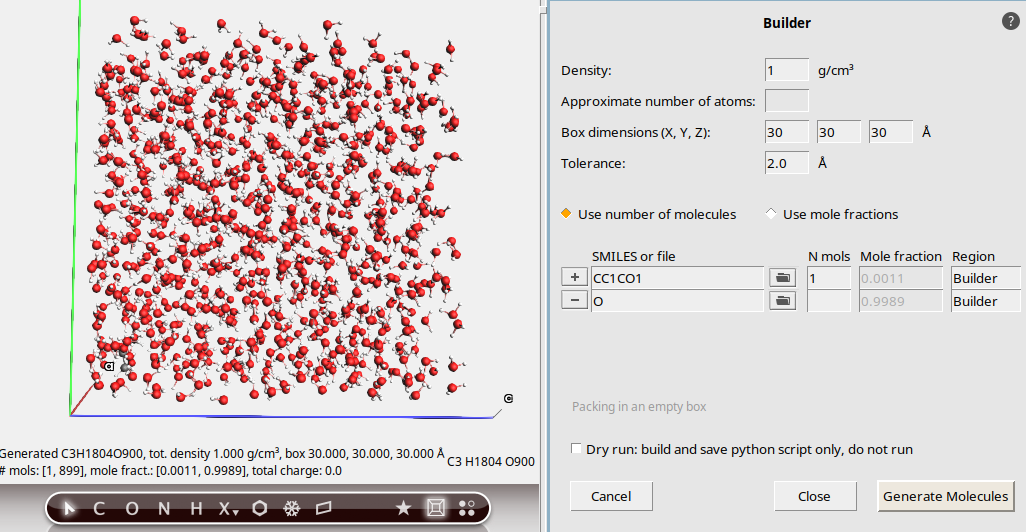
Tip
If you pack molecules that cannot reliably be generated from smiles, you may build them in AMSinput and export to .in or .xyz format. Use the folder icon in the Builder to select your exported file.
Notice that the solvent molecules are generated at random positions and orientations, with the constraint that all atoms (between different molecules) are at least the specified distance (2.0 Å) apart.
→ 
Turn on/off periodic view: View → Periodic → Periodic View or use the bottom  to activate/deactivate the periodic view tool.
to activate/deactivate the periodic view tool.
Step 2: MD equilibration¶
In this tutorial, we start with a short preparation/equilibration run.
On the right side of the AMSinput window, we already have selected the :
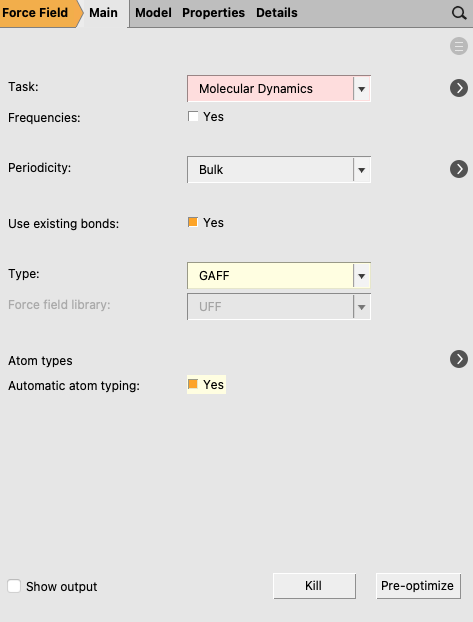
a) Configuring the duration and temperature of the equilibration trajectory¶
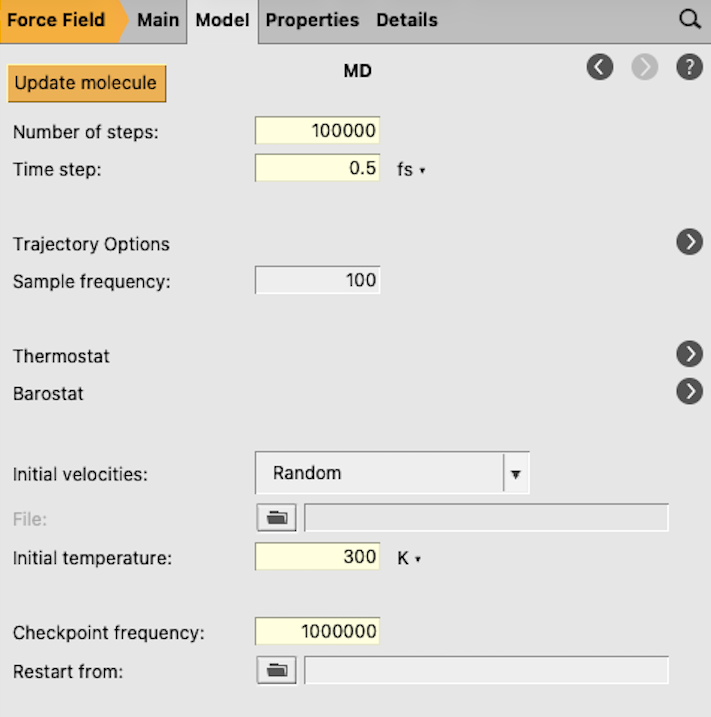
 next Task → Molecular Dynamics to go to the Model → MD panel
next Task → Molecular Dynamics to go to the Model → MD panel
The time step should not be set larger than 0.5 fs because of OH and CH bonds. In an MD run with fixed OH and CH bond lengths one could use a larger time step size, like 2 fs. For some systems one may need a larger number of steps to reach equilibrium, but that is not needed in this case.
b) Defining the thermostat for equilibration¶
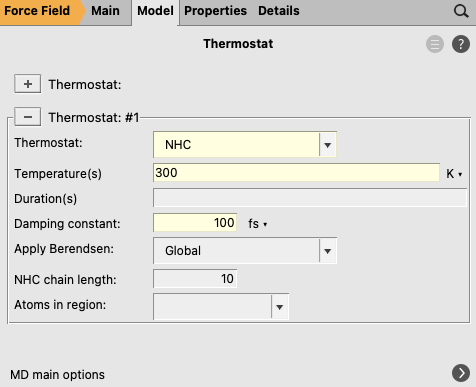
You have set an initial temperature of 300 K, but to maintain this temperature throughout the simulation it is also necessary to attach a thermostat to the system. In the equilibration step the Berendsen thermostat will be used.
next to the Thermostat to go to the Model → Thermostat panel button to add a thermostat to the simulation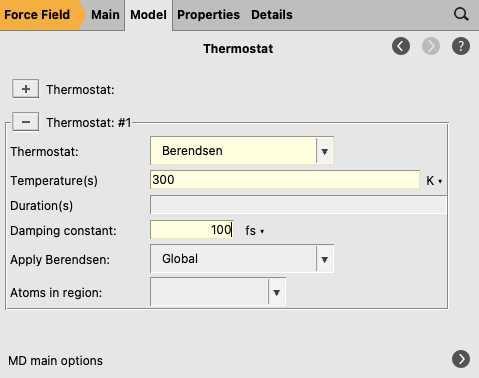
It is also possible to start with a low-temperature MD to relax the initial set-up, or to define different temperatures for the components of the system, using several thermostats for different regions. To that end, you first must define two new regions by using the Regions panel, but it is out of the scope of this tutorial, so, for your solvated 2-Methyloxirane, you will use a single thermostat.
next to MD main optionsc) Defining the barostat for equilibration¶
Just like using a Thermostat to control the temperature of the system, a Barostat can be applied to keep the pressure constant by adjusting the volume.
next to Barostat to go to the Model → Barostat panel.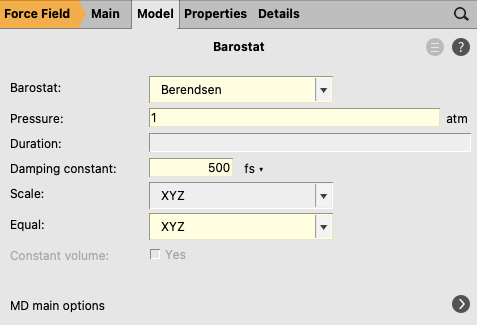 next to MD main options
next to MD main optionsd) Run the MD for equilibration¶
Now you can run your setup.
Once your job starts running, AMSjobs will show the progress of the calculation: the last few lines of the logfile:
Note that while running, the job status symbol in AMSjobs changes. If you wish to see the full logfile while the calculation is running, just click on the logfile lines in the AMSjobs window, and it will show the logfile in the AMStail window.
In the calculation first the atom types for the GAFF force field are determined, which will take some time. Next the MD steps are calculated. Let the calculation run for at least half an hour. While it is running, you can already follow its progress in AMSmovie.
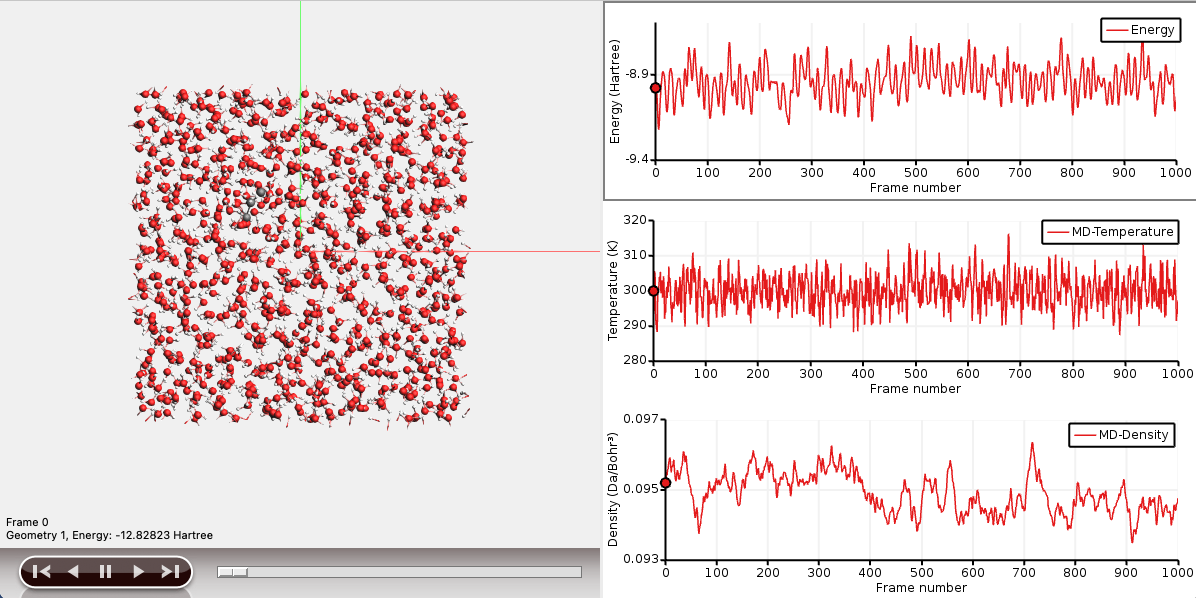
The graph on the right-hand side shows the energy as a function of the trajectory step.
You can explore different MD properties along the trajectory when it is ready or even if the simulation is still running.
Now you have two graphs. One of them is the ‘active’ graph. When you make a new graph it will always be the active graph. You can also make a graph active by clicking on it.
You can show several graphs for different properties at the same time. Temperature and Density are useful properties to evaluate the quality and convergence of your MD simulation.
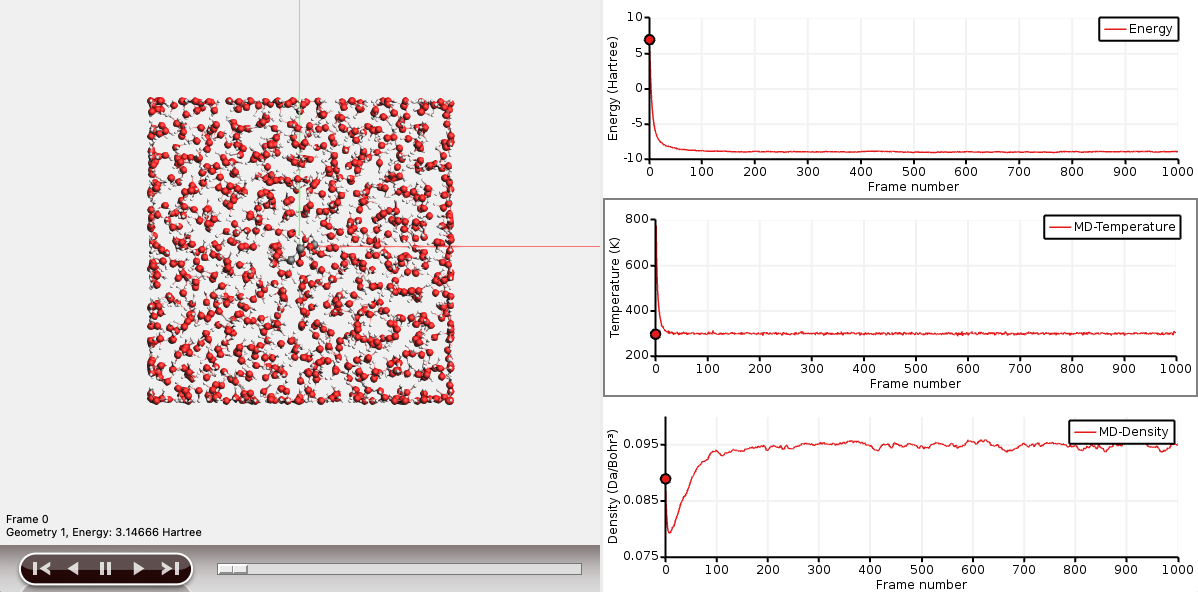
Let the calculation finish. This might take around a few hours, depending on your computer resources.
Step 3: MD production simulation¶
Here we will set up the production stage of the MD. This MD run uses the results of the equilibration run. Most setting are the same as in the equilibration run, except the settings for the thermostat. In this production run in this tutorial we will also use 100000 steps with a time step of 0.5 fs.
a) Defining the thermostat of the production run¶
b) Run the MD production simulation¶
Now you can run your setup.
Let the calculation run for at least half an hour. While it is running, you can already follow its progress in AMSmovie.
Let the calculation finish. This might take around a few hours, depending on your computer resources.
Step 4: Setting up the QM/FQ calculations¶
a) Extracting snapshots from the MD runs¶
Once your trajectory is ready, you can open it with AMSMovie and save the geometry for any snapshot.
You can also save the xyz coordinates for the entire trajectory by using File → Export trajectory as → XYZ(.xyz) but in this tutorial, we will use just two of the snapshots.
Now you can close all windows that belong to this tutorial:
AMS users should continue with the next Route # 2 to set up the QM/FQ calculations (see below).
Route # 2 (MD-experienced users and AMS users who already run the MD)
MD-experienced users can start the tutorial here if they already have at least two snapshots extracted from any trajectory.
b) Removing PBC and cutting spheres¶
This step is to be carried out if you have not removed periodic conditions yet, or if you have not cut your snapshot yet, or if you want to reduce the size of the sphere. In the AMSjobs window,
 →
→ 
c) Definition of the type of calculation, the different regions of the two-layer scheme, and their boundaries¶
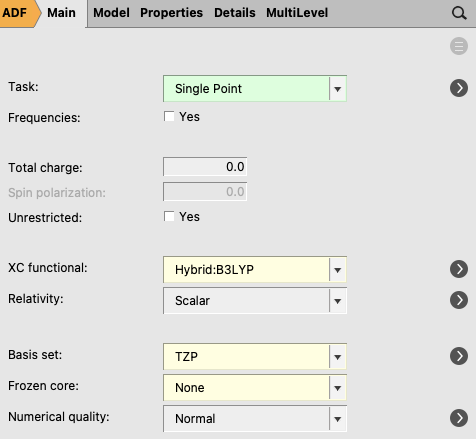
With the sphere already cut, you can proceed with the QM/FQ settings. In this tutorial you will run UV-Vis calculations, thus, in the ‘Main’ space of the panel bar,
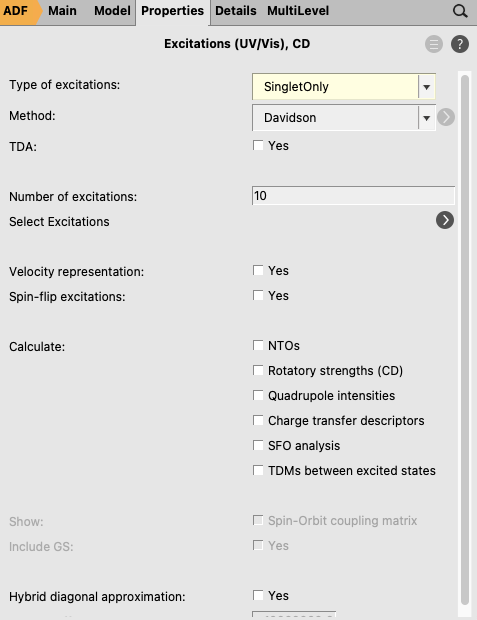
Now, using the panel bar Properties
Being a QM/MM calculation, we also need to define two regions: one for the solute, and one for the water. To set up these regions:
button to add a region button again, and rename Region_2 to Solvent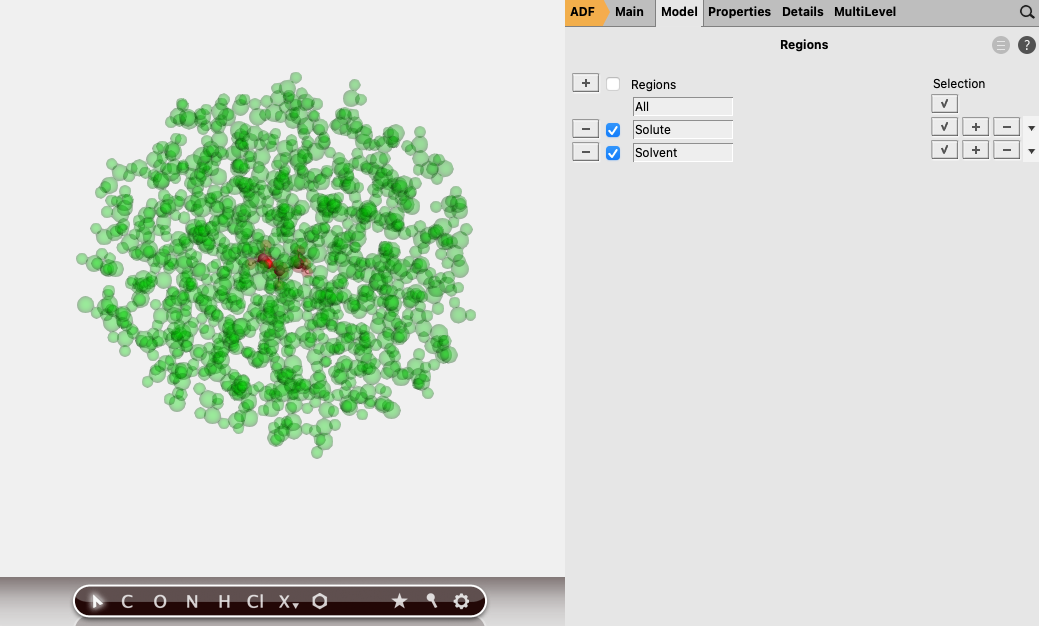
Concerning parametrization, the GUI has parameters for QM/FQ in case water is the solvent in the FQ region.
The FQ parameters (\(\chi, \eta\)) will appear in the window.
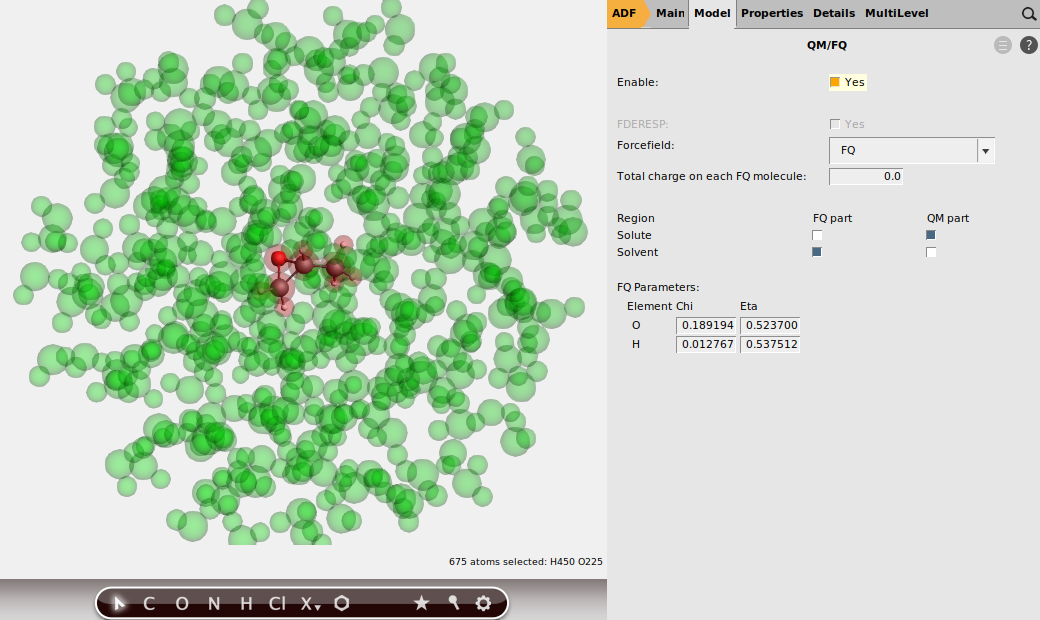
This is all the setup you need. You are now ready to run the QM/FQ calculation
d) Repeat calculation for a different snapshot¶
Step 5: Analyzing the results¶
Once the QM/FQ calculation has finished, the UV-Vis spectrum can be seen from the binary results file.
AMSspectra will start and show the calculated absorption spectrum for that specific snapshot. In the window below the spectrum, you will find a table with information. You can get more information by selecting one of the entries (or click on the peak in the spectrum), which will bring some output describing the orbitals involved.
In the table select for example the last Singlet-Singlet peak
The composition of the excitation in terms of molecular orbital transitions is listed on the right side. In many cases, you can visualize relevant orbitals (or also NTOs if you calculated them, note, however, that for hybrids one can only calculate NTOs in case of TDA) with AMSview by clicking on them in the window on the right. The active items are visually marked.
Click on the first major contribution line (with the highlighted orbitals) to bring up 2 pictures of the orbitals, one of the occupied orbital with red and blue lobes and one of the virtual ones with turquoise and ochre lobes.
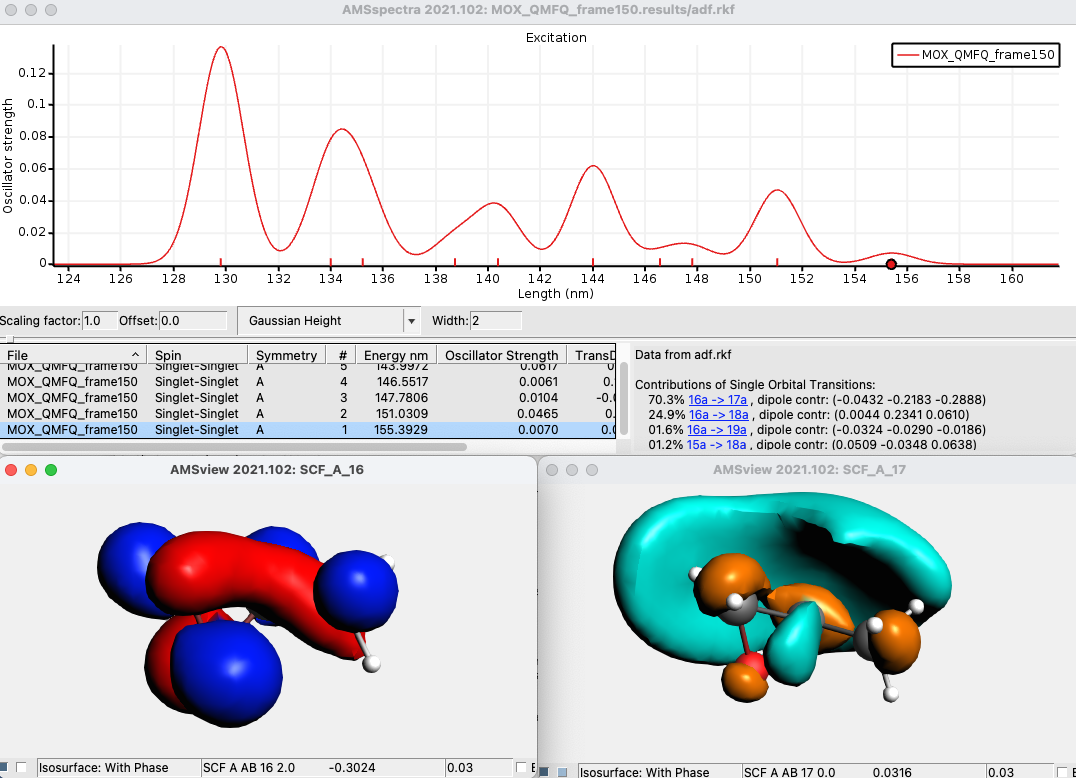
Close the two windows showing the orbitals.
Next, if you calculated mote than one snapshot.
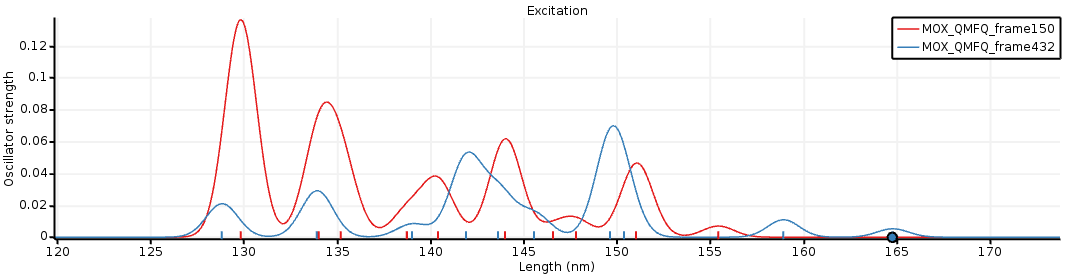
Next an averaged spectrum is calculated.
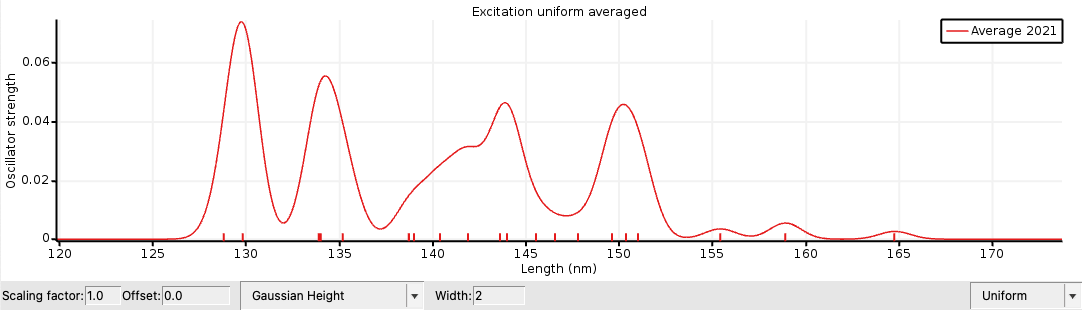
You can see the individual spectra if one selects View → Average/All Curve. Note that at the moment the excitation spectra are artificially broadened (Width parameter). This broadening can be estimated in a more physical way if one calculates the average spectrum of excitation energies for many MD snapshots.