TD-CDFT Response Properties For Crystals (OldResponse)¶
BAND can calculate response properties such as the frequency-dependent dielectric function within the theoretical framework of time-dependent current density function theory (TD-CDFT).
This introductory tutorial will show you how to:
Set up and run a BAND single point calculation (using AMSjobs and AMSinput)
Set up and run a BAND TD-CDFT, linear response calculation (using AMSjobs and AMSinput) with the
OldResponsemethodVisualize the dielectric function using AMSspectra
If you are not at all familiar with our Graphical User Interface (GUI), check out the Introductory tutorial first.
Step 1: Create the system¶
We now want to create a silicon crystal. Let us import the geometry from our database of structures:
 →
→ 
 .
.
Step 2: Run a Single Point Calculation (LDA)¶
We will first perform a single-point calculation of our Silicon crystal using LDA.
Tip
It is good practice to do a convergence study w. r. t. k-space sampling and basis set.
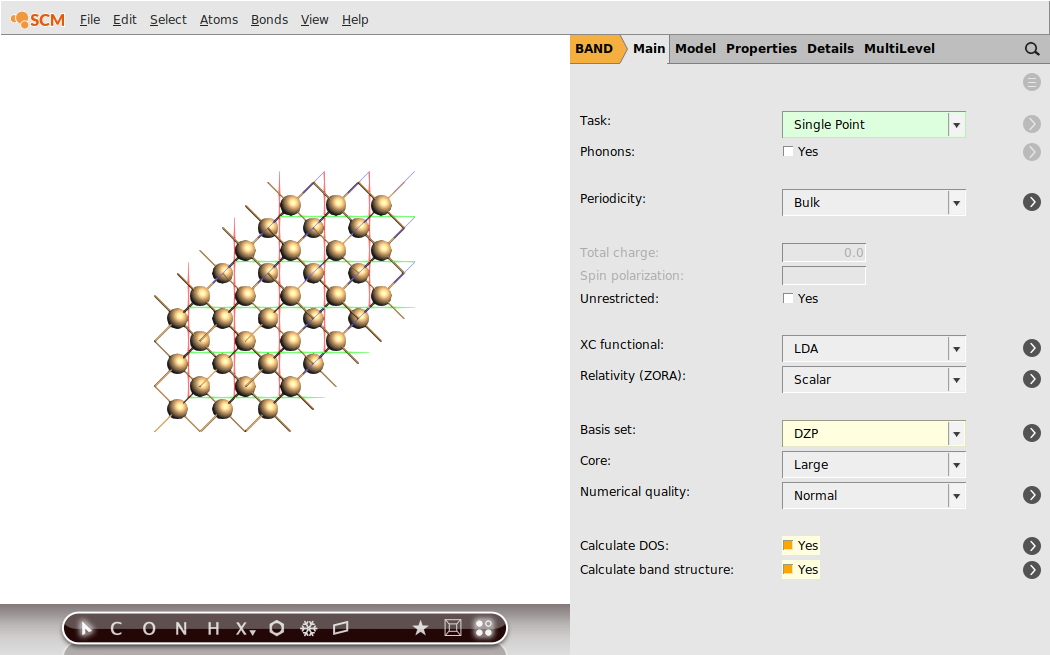
After the calculation finished, we can check the band-gap energy e.g. in the logfile. Furthermore, with the help of the AMSbandstructure module we can validate that a very basic property is reproduced by this rather poor k-space sampling - Si diamond has an indirect band-gap.
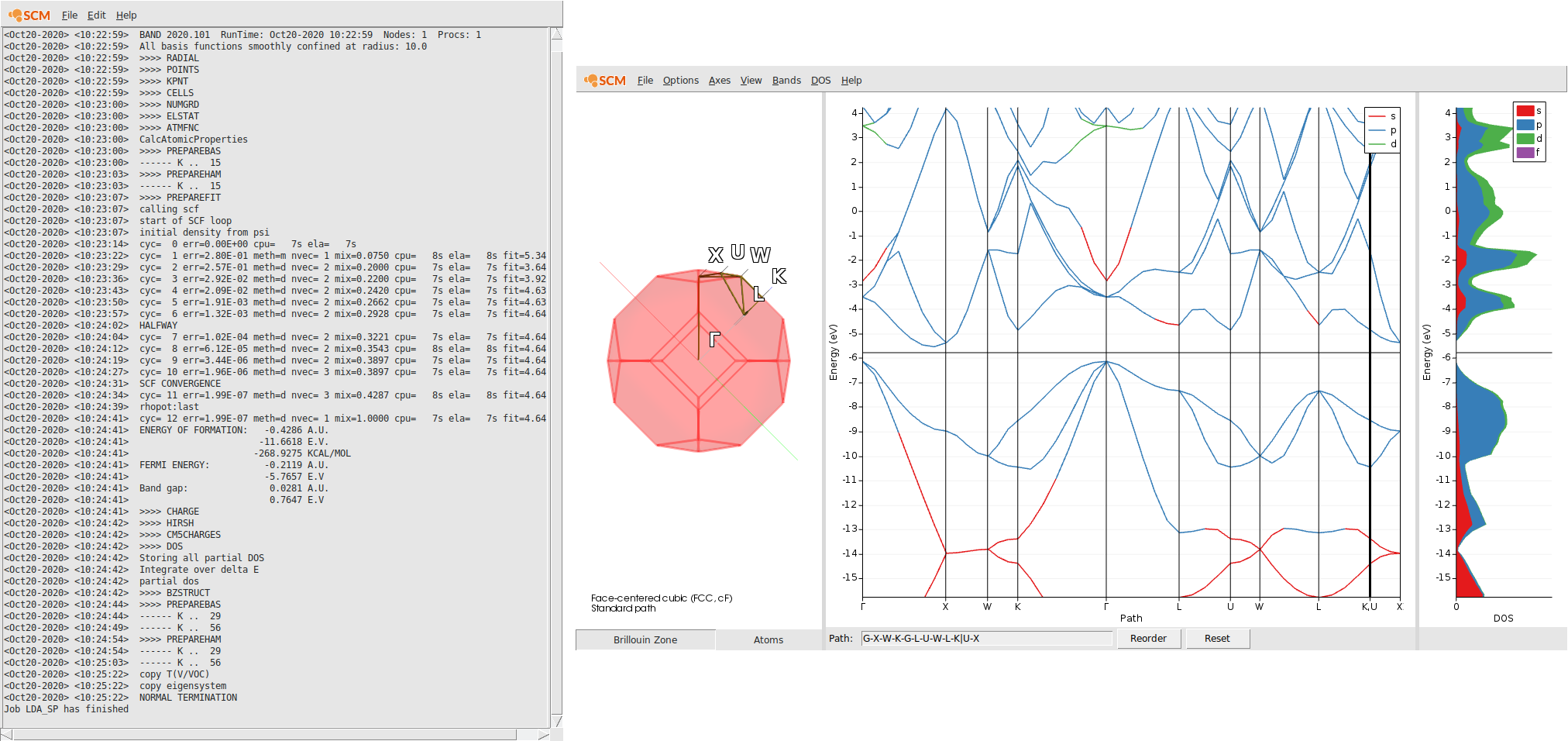
Step 3: Run an OldResponse Calculation (ALDA)¶
We will now calculate the frequency-dependent dielectric function using linear response TD-CDFT.
In the previous step we learned that the calculated band-gap for the chosen theoretical level is 0.76 eV. This is 0.35 eV below the experimental band-gap. Hence, we will shift the virtual crystal orbitals by this value in energy space. We will sample the frequency range from 2.0 eV to 5.0 eV with a step-size of 0.1 eV.
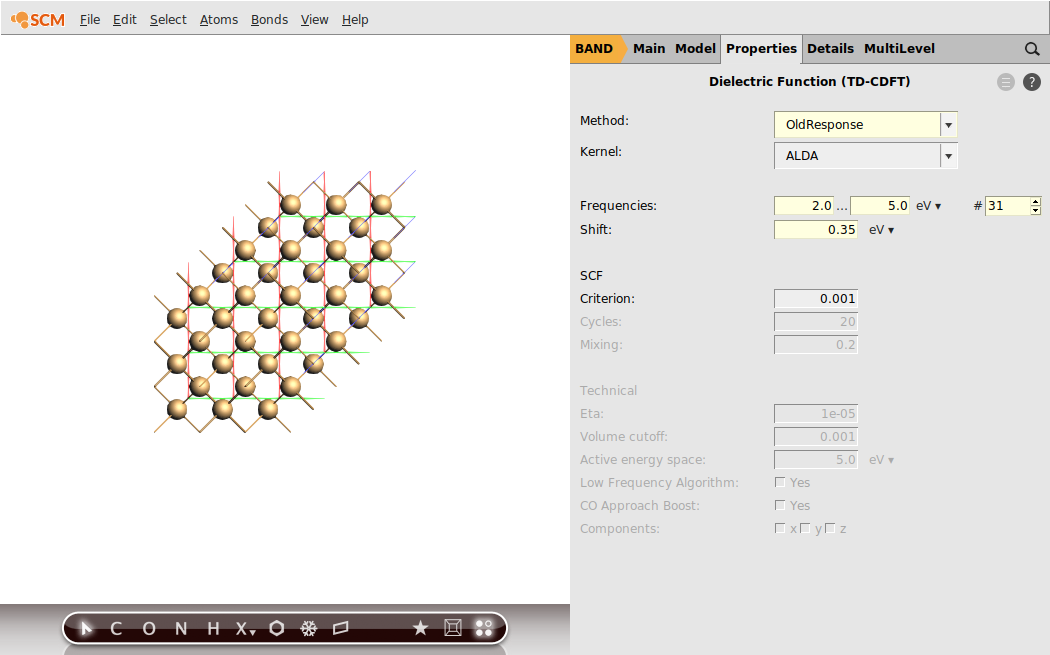
After the calculation finished, we can visualize the dielectric function using AMSspectra.
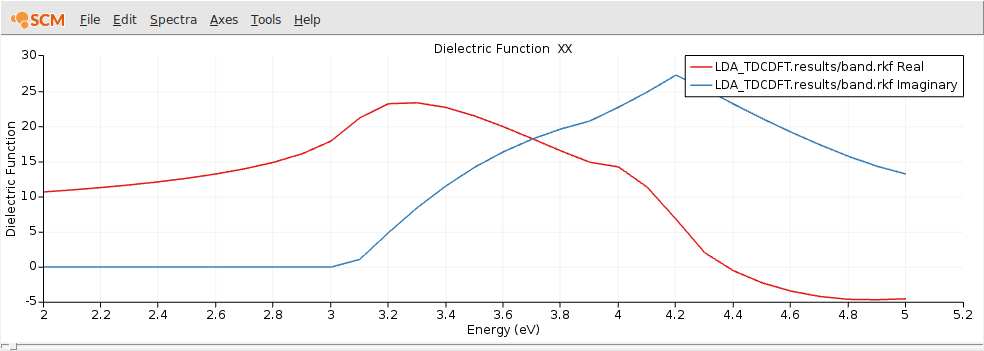
The general features of the frequency-dependent dielectric function are nicely reproduced, but for a quantitatively better result one has to converge the k-space sampling, basis set and numerical integration. Also switching to the Berger2015 kernel would improve the results further. [Ref]