Vibrational progression of an OLED phosphorescent emitter¶
Triplet harvesting in OLEDs by transition metal complexes increases the maximum efficiency from 25% to 100%. Singlet excitons are rapidly converted to triplet excited states and fast phosphorescence can be achieved by strong spin-orbit coupling.
In this example we calculate the frequencies of the T1 and S0 state of Pt(4,6-dFppy)(acac) in the gas phase to determine the vibronic fine structure from the Franck-Condon factors. We compare these with the experimental results from the Yersin group, who studied Pt(4,6-dFppy)(acac) in n-octane. [1]
The sample command line input files can be downloaded here.
To calculate the overlap of the vibronic wave functions, we first need to calculate the frequencies of the two electronic states involved, followed by the Franck-Condon calculation. So we have three steps:
Optimize the lowest singlet state (S0) and calculate frequencies
Optimize the lowest triplet state (T1) and calculate frequencies
Calculation of the Franck-Condon Spectrum
You can start straight away with the example input files or read further to learn how to set up these three calculations in the GUI for your own complexes.
1. Optimize lowest singlet state (S0)¶
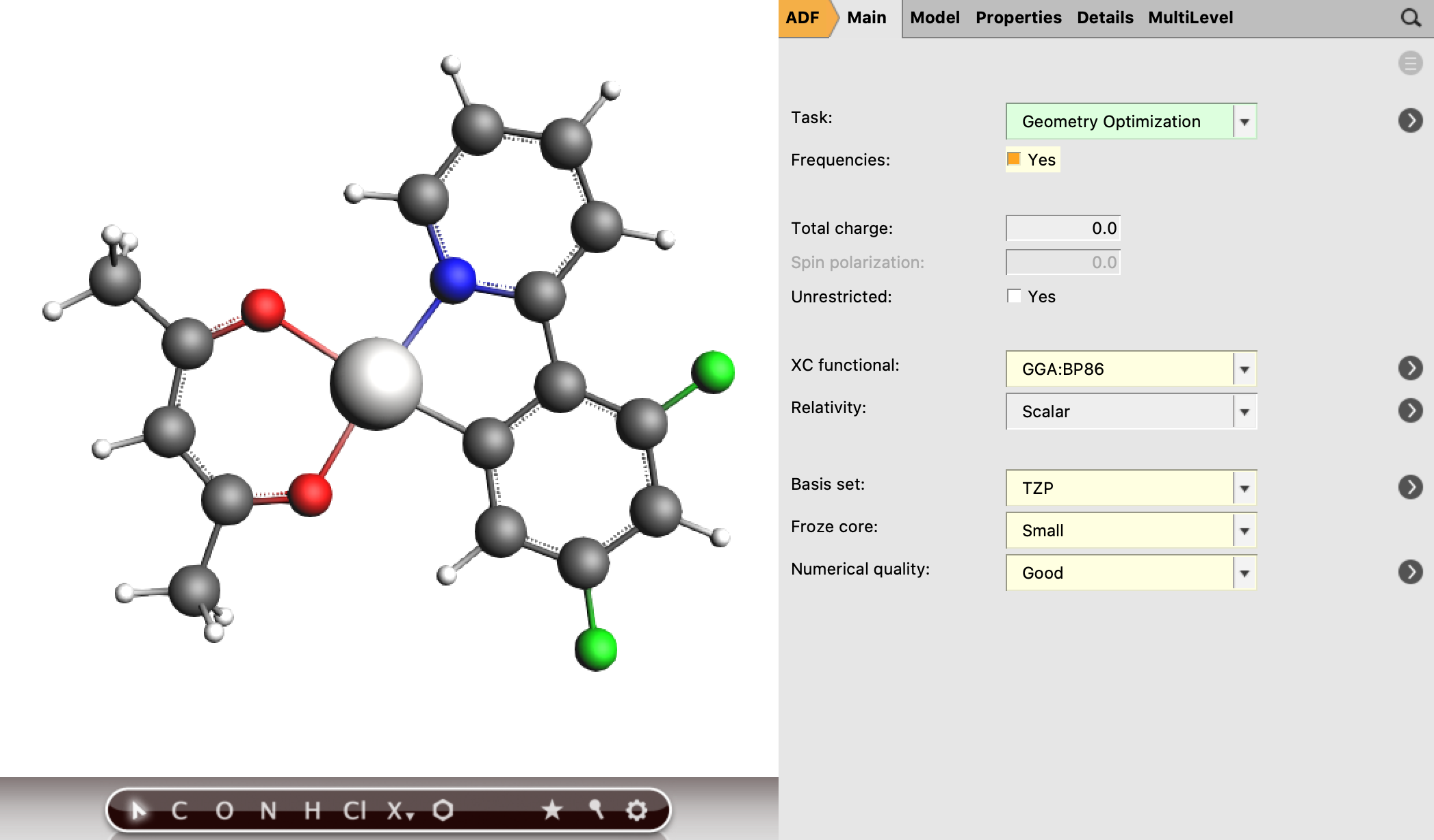
First, the geometry of the complex (in its lowest singlet state) must be optimized by performing the following steps. (Another basis set and functional can be used as well.)
Remark: Pt(4,6-dFppy)(acac) has Cs-symmetry. The use of symmetry may speed up the calculation and may improve the analysis of the results.
xyz coordinates
2. Optimize lowest triplet state (T1)¶
For calculating the complex in its T1 state, the same DFT settings should be applied as for S0, but an unrestricted calculation needs to be performed.
Remark: An open shell electronic configuration may break symmetry. In this case Pt(4,6-dFppy)(acac) also has Cs-symmetry in the lowest triplet state, as one may check if one looks at the results of the frequency calculation. In general, however, one may have to break symmetry in the starting geometry, in order to get a non-symmetric optimized geometry.
xyz coordinates2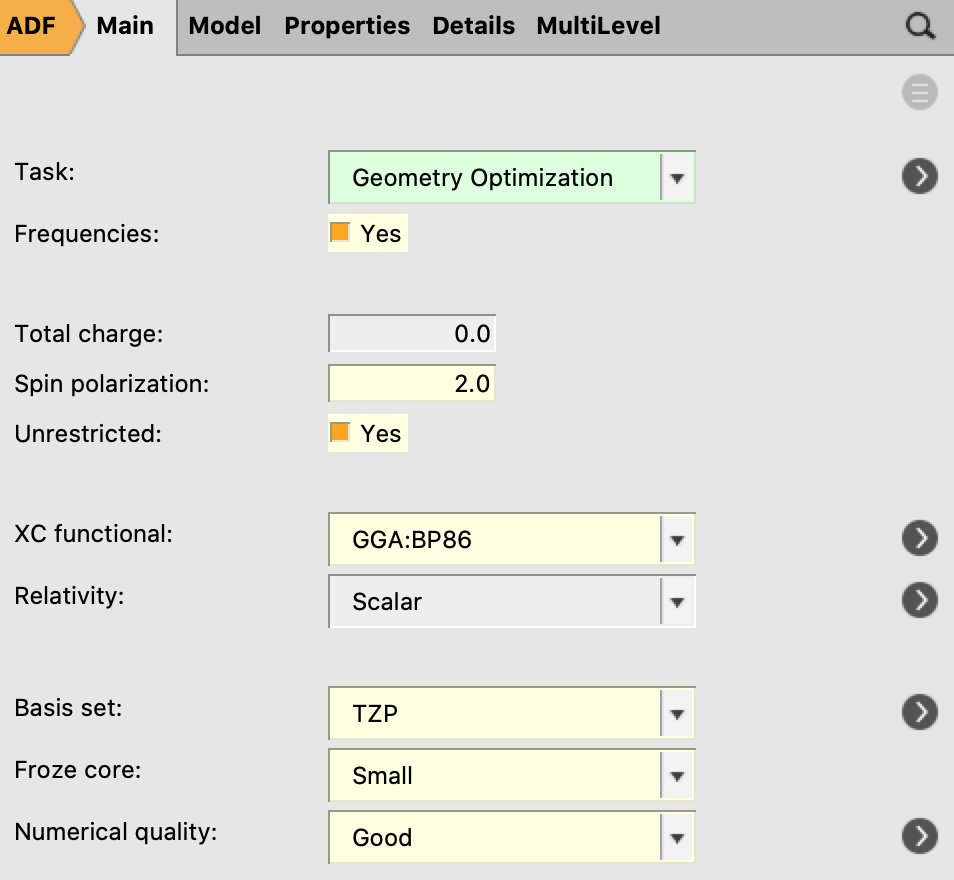
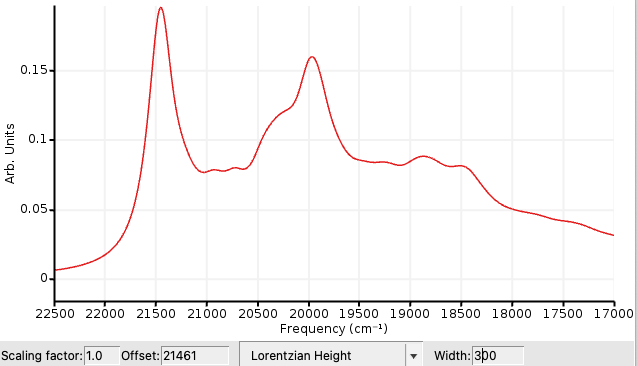
This difference in energy should be about 2.5 eV which is in reasonable agreement with the experimental result for the 0-0 transition, which is 21461 cm-1 (2.66 eV) for Pt(4,6-dFppy)(acac) in n-octane. [1].
3. Calculation of the Franck-Condon Spectrum¶
Next the Franck-Condon spectrum will be calculated. Important is to make a new AMSinput file. In this example we calculate the vibrational resolved spectrum for emission from the T1 state to the S0 ground state. We do not consider vibrational excitations in the T1 state (i.e. “hot states”). Thus the reference state is the vibrational ground state of the electronically excited T1 state.
S0_GeoFreq.results)adf.rkf file from the T1 calculation (e.g. in the T1_GeoFreq.results folder)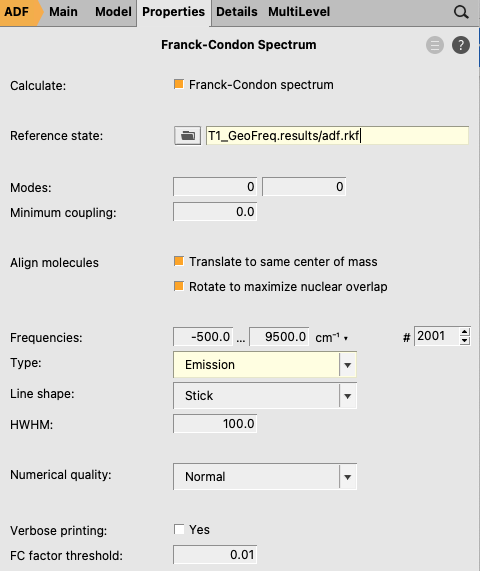
More information about the calculation of Franck-Condon spectra can be found in the AH-FC: Adiabatic Hessian Franck-Condon section.
The output will list the spectral intensity from -95000 cm-1 to 500 cm-1 (relative to the 0-0 transition) by taking into account the overlap of the vibronic wavefunction (Franck-Condon factors). The FCF spectrum can be visualized at SCM → Spectra. The lines can be Gaussian-broadened to take into account thermal broadening.
300 for the Width21461 (experimental 0-0 transition) for the Offset17000 for Minimum value and 22500 for Maximum value