Proton affinities with DFTB3¶
In this tutorial we will use the DFTB engine to calculate a proton affinity, defined as the negative gas phase enthalpy for the protonation reaction A- + H+ → AH. Gaus, Cui and Elstner have shown that third-order terms (DFTB3) generally improve the proton affinities with respect to second order self-consistent charges (SCC).
We will calculate the proton affinity of the acetate anion, CH3 COO- . In the first step of this tutorial, we will optimize acetic acid at the DFTB3 level. The second step will perform the computation on the anionic species, and compute its proton affinity.
Step 1: Optimization of the neutral molecule¶
 →
→ 
Now we need to obtain an acetic acid molecule and set up a geometry optimization at the DFTB3 level.
ctrl+r. You will be asked to give your job a name.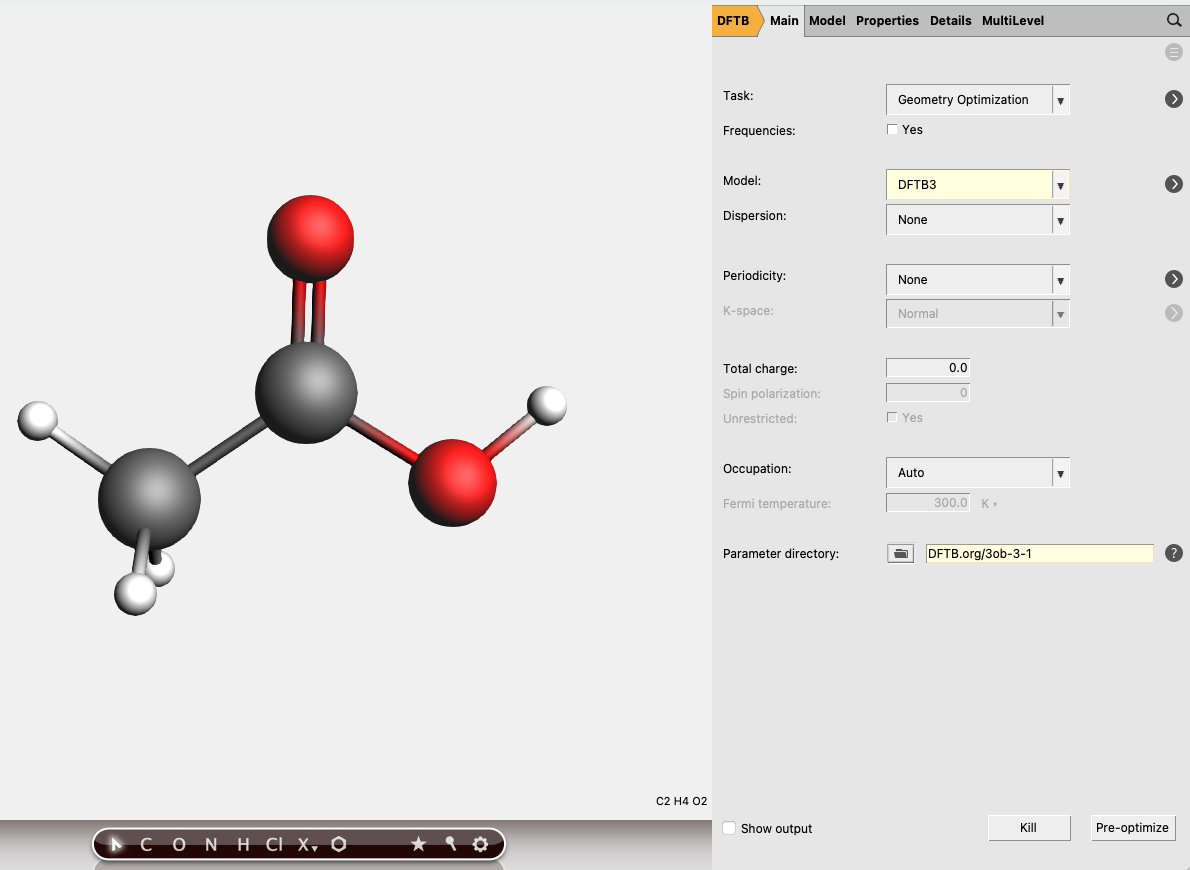
When the optimization is complete (it should not take longer than a second), one can click on the ‘Update molecule’ button that will pop up in AMSinput window to update the geometry. For this tutorial it does not matter if you load the new geometry or not, as we are not going to run any calculations with the optimized geometry.
The total energy of the acetic acid molecule should be close to E(HAc) = -11.5755 Hartree. In order to calculate the proton affinity we now need to detach a proton and calculate the energies of the acetate ion and the lone proton.
Step 2: Optimization of the acetate and the hydrogen ions¶
In order to perform the calculation on the acetate ion, we will remove the hydrogen ion from the previously computed acetic acid molecule.
ctrl+r to run it.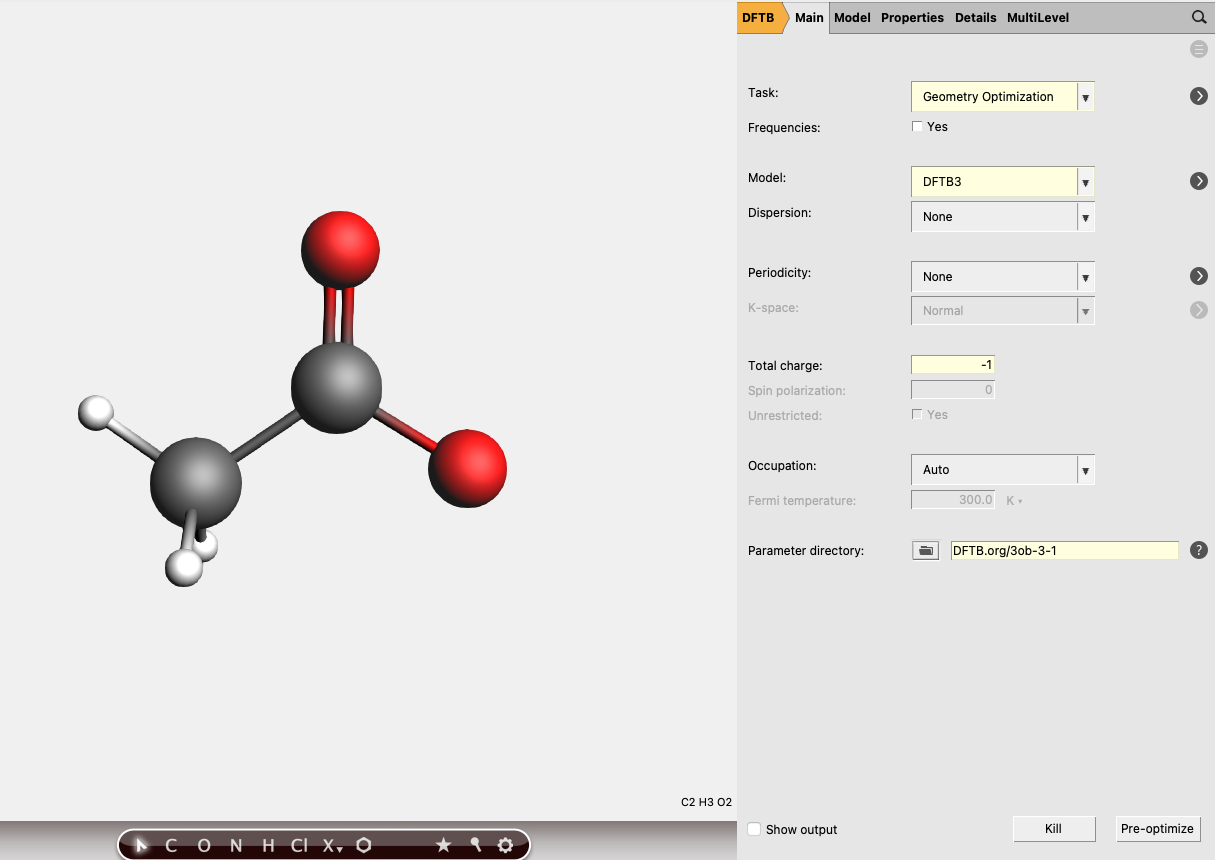
You should obtain an energy of about E(Ac- ) = -11.2458 Hartree for the acetate ion.
Finally we also need the energy of a lone proton. There is no point in performing a geometry optimization for a single proton, so here we simply perform a single point calculation.
ctrl+r to run it.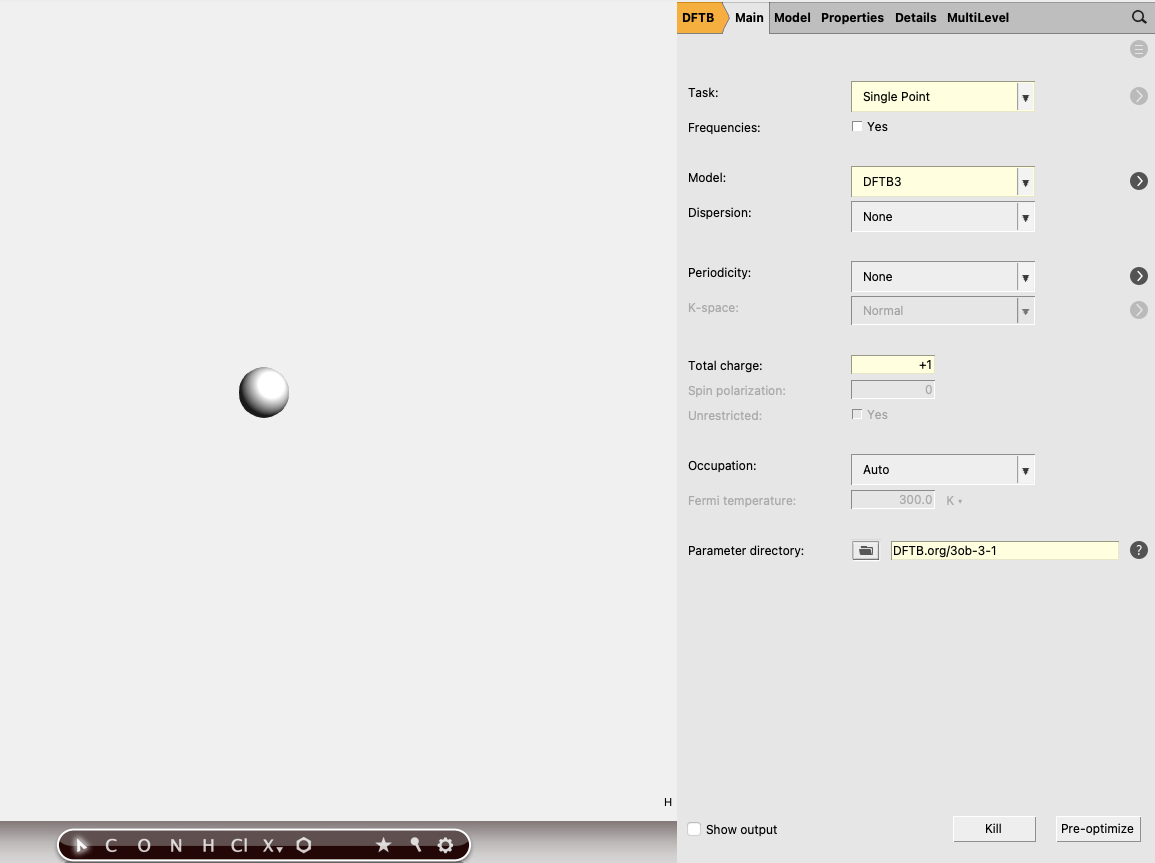
The total energy of the proton should be close to E(H+ ) = 0.2407 Hartree.
The proton affinity is computed as PA = E(Ac- ) + E(H+ ) - E(HAc), resulting in a final proton affinity of 0.5704 Hartree, or 357.93 kcal/mol.
We leave it as an example to calculate the PA with DFTB2 (SCC-DFTB), and compare it with the high-level ab initio results that are also quoted in the DFTB3 paper.