Solvents, environments and multi-layer (QM/MM) calculations
The effect of the environment on the reactivity or spectroscopic property of a central molecule or moiety can be substantial.
In the Amsterdam Modeling Suite there are various options to treat such environment effects with different levels of accuracy and efficiency. Besides implicit solvent models, there are embedding methods and multi-layer methods which take into account the environment.
Also see the tutorial on how to add explicit solvent molecules in a 3D box.
Implicit solvation: COSMO and SM12
The polarizable continuum model COSMO is a quick and efficient method to include solvation effects implicitly in ADF and in BAND. It can be applied to (excited state) geometry optimizations and a range of spectroscopic properties. The generalized Born method SM12 is only available for single point energy calculations. Implicit solvation for molecular systems with DFTB is available through the GBSA generalized Born method.
Both COSMO and SM12 can also be applied to periodic systems in our periodic DFT code BAND. There is also a fast local COSMO version for large systems with, in combination with the DFT-in-DFT method FDE.
Frozen-Density Embedding (FDE)
 Essentially a divide-and-conquer method dividing the full system in smaller systems (subsystem DFT). While calculating the density of one subsystem, the density of the others are kept frozen. This can be done in freeze-thaw cycles to self-consistently relax the total density of the system in a way that scales favorably – linear with the number of subsystems – and thus applied to really large molecules or systems such as many solvation shells.
Essentially a divide-and-conquer method dividing the full system in smaller systems (subsystem DFT). While calculating the density of one subsystem, the density of the others are kept frozen. This can be done in freeze-thaw cycles to self-consistently relax the total density of the system in a way that scales favorably – linear with the number of subsystems – and thus applied to really large molecules or systems such as many solvation shells.
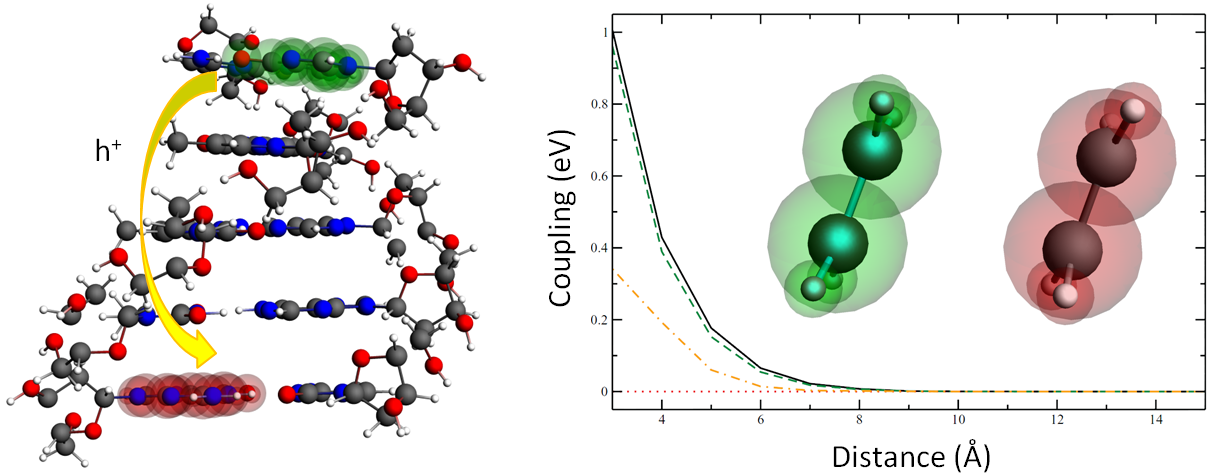
FDE gradients are available, and FDE also enables the calculation of a range of important properties especially for organic electronics (excitation energies, electron transfer, exciton transfer, charge generation and recombination).
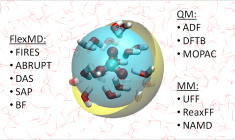
Multi-layer calculations: QM/MM, QM/QM, DIM/QM
From AMS2020 onwards, there is central hybrid engine, for classic QM/MM as well as multi-layer calculations with any combination (‘ONIOM’, used to be available via QUILD). These hybrid, multi-layer calculations can be run for molecules as well as periodic systems (1D, 2D, 3D).
You can use electrostatic embedding (additive scheme) only for two-layer QM/MM calculations with ADF, BAND or DFTB for the QM layer, and Tripos, Amber, or UFF for the Force Field.
With mechanical embedding, you can use any number of layers, any type of engine (various QM, MM, ML potential, ReaxFF) combination, and any periodicity for your multi-layer calculations.
FlexMD can also be used to set up subtractive QM/MM calculations and it can deal with many internal and external codes. It also enables adaptive QM/MM methods to deal with water molecules crossing the QM/MM boundary. For FlexMD you would also need to be able to describe your active QM system with the force field.
DIM/QM is a specialized QM/MM approach that includes polarizable force fields (DRF, see tutorial) to take into account solvation effects on opto-electronic properties of the central molecule, and also polarizable interaction models to deal with molecules on metallic nanoparticles, including many plasmon-induced non-linear optical properties (SEROA, SEHRS, PETPA)
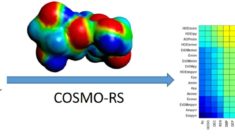
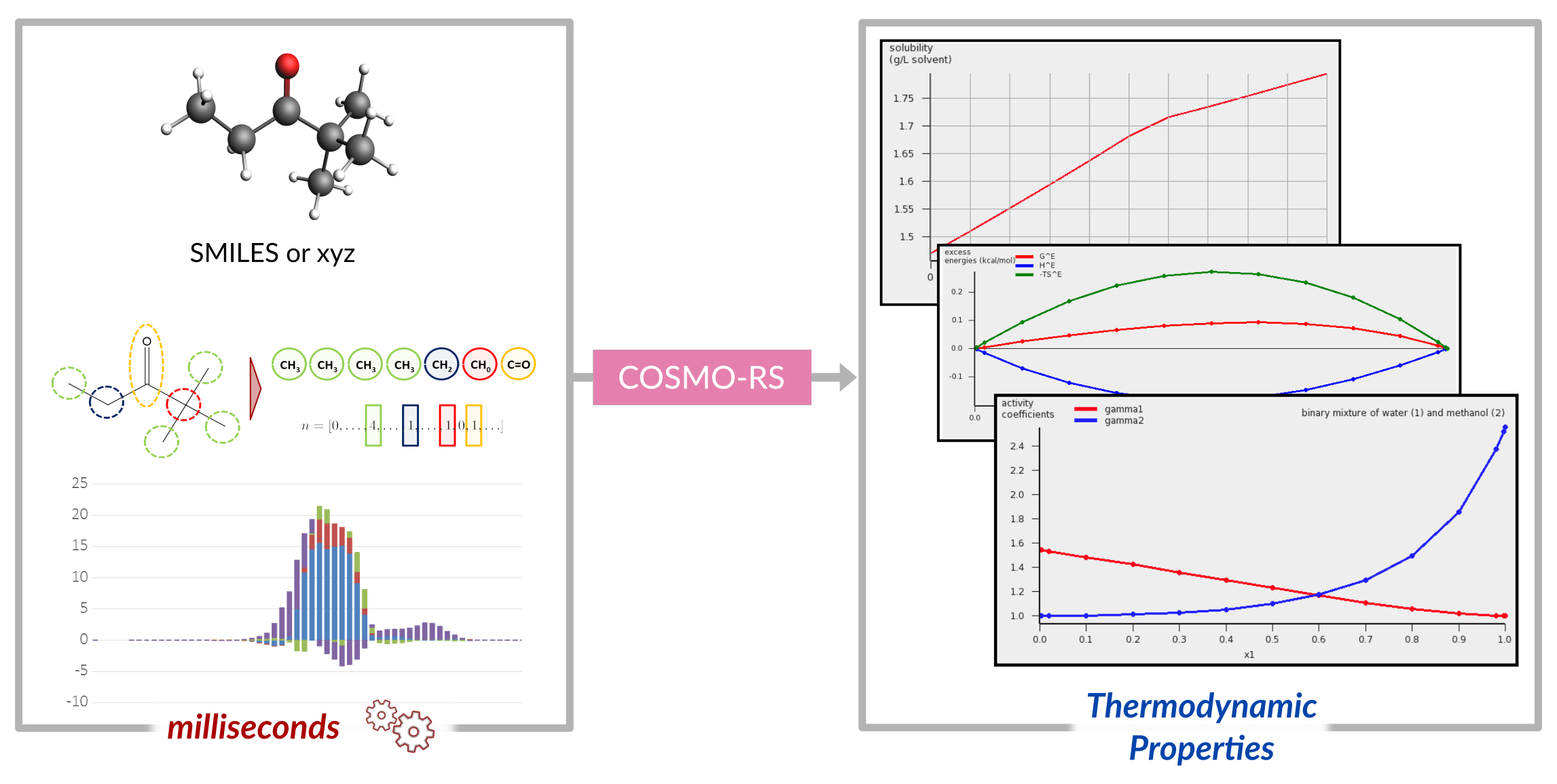
COSMO-RS
Is not the same as COSMO, but it uses molecules calculated with a specific COSMO protocol to calculate thermodynamic properties in solution. The ‘Realistic Solvent’ extension uses the surface charges and a few other terms to generate pseudo-activity coefficient which form the basis for many accurate properties, such as solvation free energies, solubilities, vapor pressures and partition coefficients. Similarly these properties can be calculated through the COSMO-SAC model and recently faster property prediction methods have been implemented (UNIFAC and QSPR-type predictions)
3D-Reference Interaction Site Model (3D-RISM)
Expert option which embeds the central system in the potential of an averaged solvation configuration, usually based on a force field. 3D-RISM can yield accurate thermodynamics and solvent shifts on optical properties.
SCRF and VSCRF
The self-consistent reaction field is based on the Poisson–Boltzmann equation to self-consistently optimize the quantum charges and those of the solvent surroundings (polarizable force field). It can be combined with a delta-SCF type approach to get vertical excitation energies (VSCRF)