Molecular dynamics: Water on an aluminum surface¶
This tutorial will help you to:
- create an aluminum slab
- add a solvent (water)
- set different thermostats (temperatures) for different regions of the system.
- run the molecular dynamics simulation and see what happens
For this simulation we will use the ReaxFF force field through the AMS driver program. In ADFinput this combination is called the ReaxAms module. Note that the (reactive) force field is identical to what is used in the stand-alone ReaxFF program.
Note
The purpose of this tutorial is to demonstrate how to technically set up and run a simulation. The employed Al-H2O ReaxFF force field was parameterized to handle similar, but not exactly the same, types of systems as in this tutorial. Always do your own testing to see if the results from a simulation are reasonable.
Step 1: Start ADFinput in ReaxAms mode¶
- 1. Start ADFjobs2. Use the SCM → New input menu command3. Switch to ReaxAms:
 →
→ 
Step 2: Creating the surface¶
Bulk aluminum has an fcc crystal structure, with a lattice constant of 4.05 Å of the conventional unit cell.
To create the surface, we first build the primitive unit cell of bulk aluminum:
- 1. Edit → Crystal → Cubic → fcc2. In the dialog that appears:3. Select the preset Al4. Click the OK button5. View → Periodic → Repeat Unit Cells
In your molecule editor screen you should see a picture of the bulk aluminum structure. The primitive unit cell contains one atom. As ReaxFF input by default does not show repeated cells, it had to be turned on explicitly. Otherwise you see only the single atom in the unit cell.
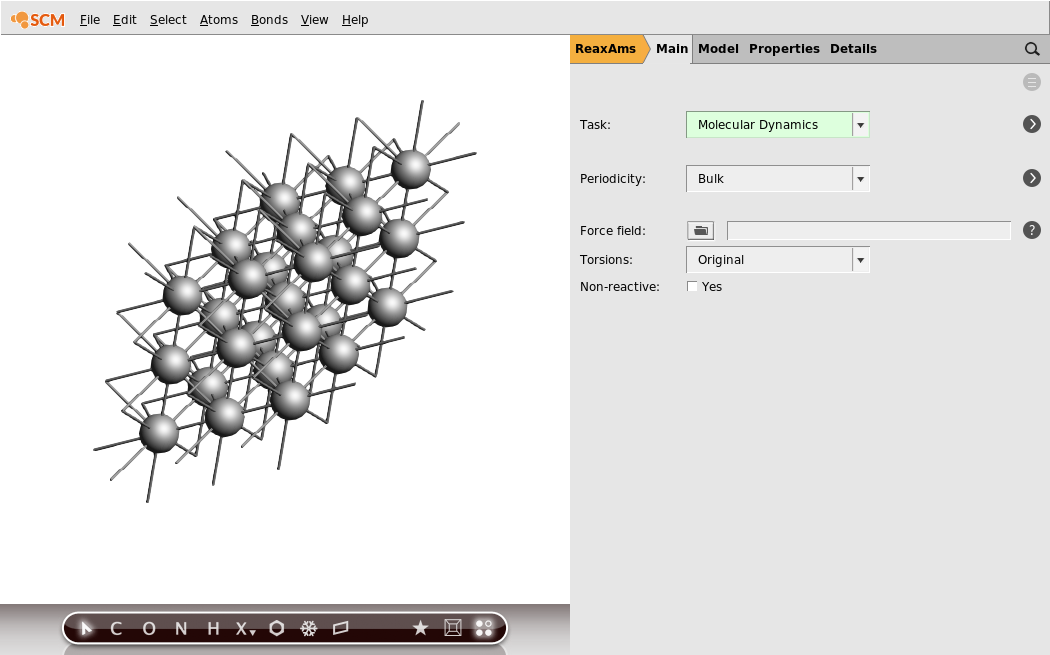
Tip
In the menu bar, select View → Properties (including estimated) to see some properties of the system, such as stoichiometry, angles between lattice vectors, and the system density.
Now create the surface. Surfaces are constructed by specifying the Miller indices, which requires that the conventional (and not primitive) cell be used.
- 1. Edit → Crystal → Primitive To Conventional Cell. This creates a conventional cubic unit cell with lattice constant 4.05 Å.2. Edit → Crystal → Generate Slab…3. Set the Miller indices to
0,0,1.4. Set the number of layers to25. Click Generate Slab6. Rotate to see the system from the side
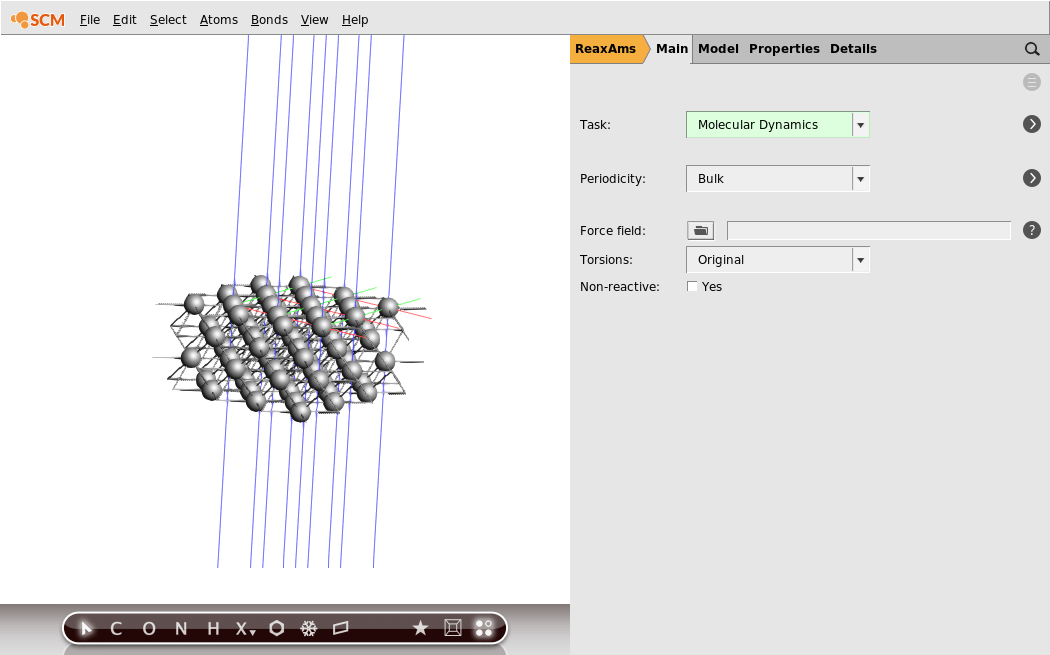
You should be able to see it is a four-layer slab.
In this case we do not want just one unit cell, but a much bigger piece of the slab:
- 1. Turn off periodic view: View → Periodic → Repeat Unit Cells2. Edit → Crystal → Generate Super Cell…3. Put
10,10,1on the diagonal4. Click OK5. Rotate the system and get a better view
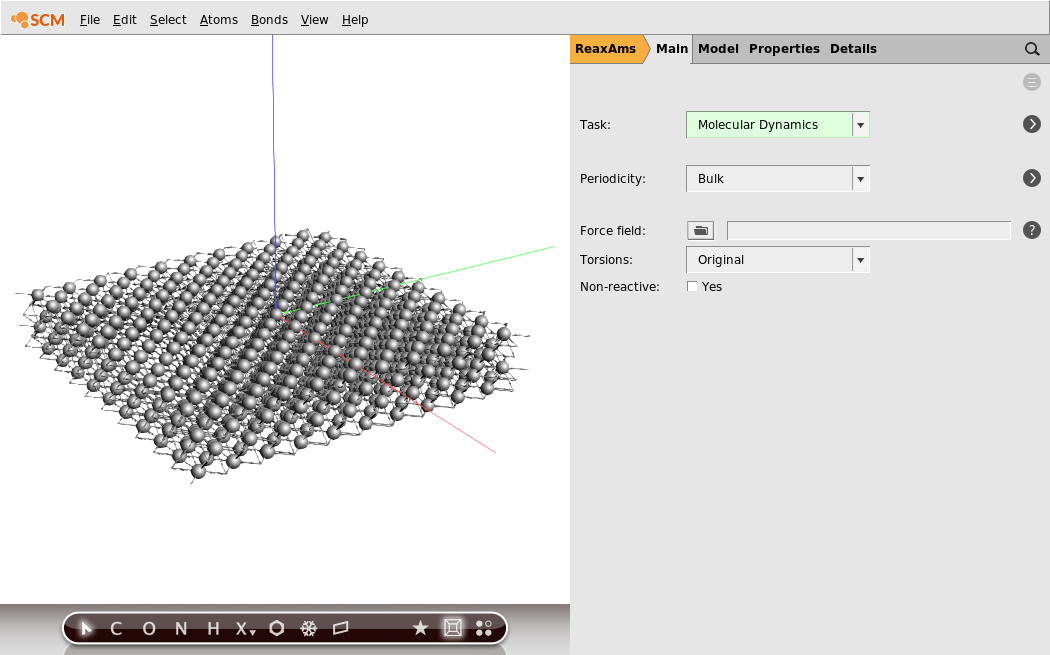
Now we have a slab of aluminum, four layers thick.
Step 3: Add solvent¶
The next step is to add water to the system.
- 1. Edit → Builder2. Change the third dimension of the lattice vectors to
50.03. Typewain the line with ‘Fill box with’4. Select Water (ADF) from the search results5. Specify1600copies.7. Click the Generate Molecules button on the bottom8. Click View → Periodic → Show Unit Cell to visualize the box9. Rotate to your favorite view
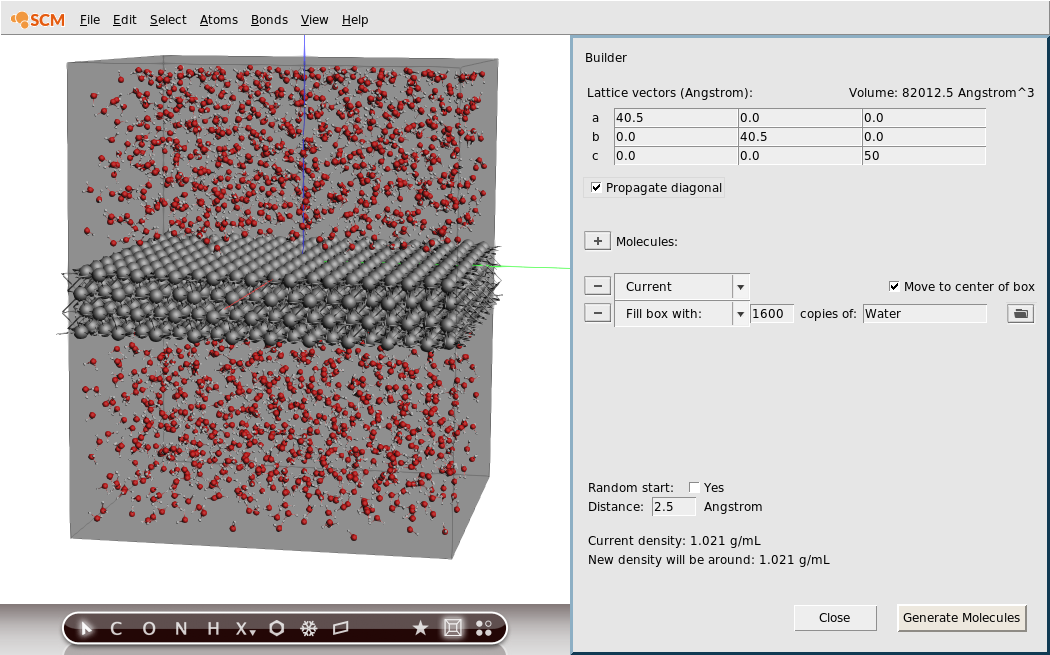
Note
After the water has been added, the density \(\rho_{\textrm{tot}} = 1.021\) g/mL. This is the density of the entire system, including the Al slab. The resulting density of the added water can be estimated as \(\rho_{\textrm{water}} = \frac{m_{\textrm{tot}}-m_{\textrm{Al}}}{V_{\textrm{tot}}-V_{\textrm{Al}}} = \frac{\rho_{\textrm{tot}}V_{\textrm{tot}} - \rho_{\textrm{Al}}V_{\textrm{Al}}}{V_{\textrm{tot}}-V_{\textrm{Al}}} = \frac{10^{-24}}{10^{-24}}\cdot\frac{1.021\cdot82012.5-2.97\cdot4.05^3\cdot2\cdot10\cdot10}{82012.5-4.05^3\cdot2\cdot10\cdot10} = 0.64\) g/mL. In this tutorial, this lower density (compared to liquid water) is used because the water will be heated to a very high temperature (see Step 4)
Step 4: Set up the simulation, including a temperature regime¶
Now we will set up the MD-simulation. We will use the Al-water force field and a Nose-Hoover thermostat with a default damping constant of 100 fs:
- 1. Close the Builder by clicking the Close button on the bottom2. Click on the Force field folder icon and select the Al-H2O force field3. Make sure the Task is set to Molecular Dynamics and click
 to go the Model → MD panel4. Set the Number of steps to
to go the Model → MD panel4. Set the Number of steps to10000005. Set Initial velocities to Zero6. Go to the Model → Thermostat panel by clicking the next to Thermostat7. Click the + button to create a new thermostat8. Set Thermostat to NHC9. Set Damping constant to100.0fs
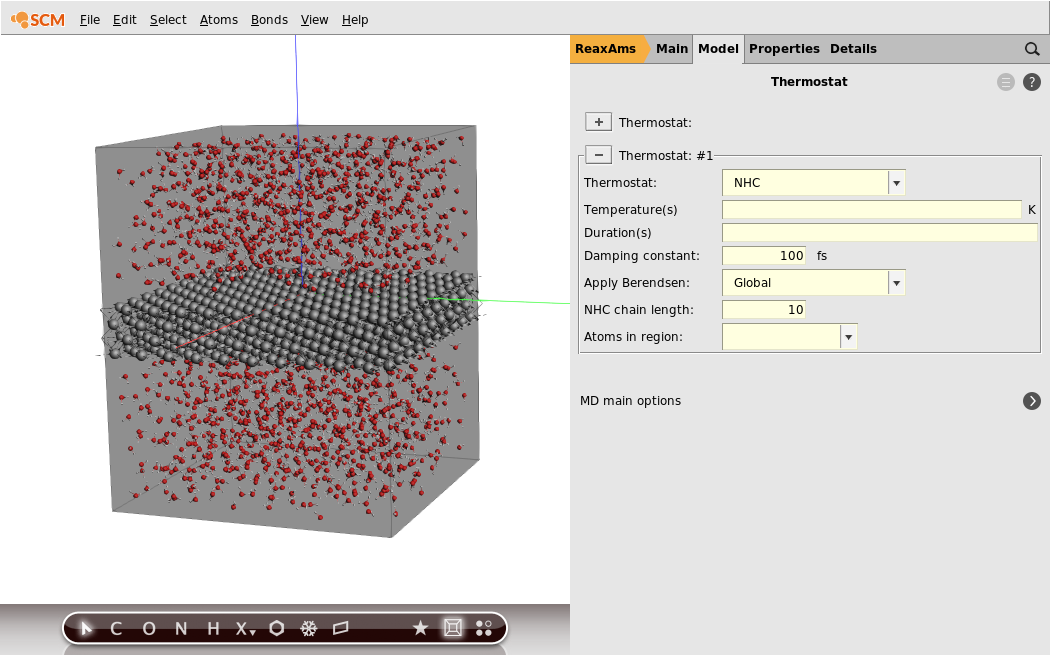
For the purpose of this tutorial we want to quickly see something happen in our simulation. We will therefore use a very high temperature for the water, but try to keep the aluminum cool. Also, we will start with a low temperature MD to relax the initial set-up. This can all be accomplished using several thermostats for different regions.
For this we first need to define two new regions: one for the aluminum slab, and one for the water. In ADFinput, regions are just defined as a collection of atoms, which can be set up via the Regions panel:
- 1. In the panel bar, click Model → Regions2. Click the button with the checkmark under Selection for the
Auto_Generatedregion
By pressing the select button you have selected all atoms in the Auto_Generated region. This region will always contain the atoms that are added by the Builder tool we used earlier. Thus, as you can see, all water molecules are selected.
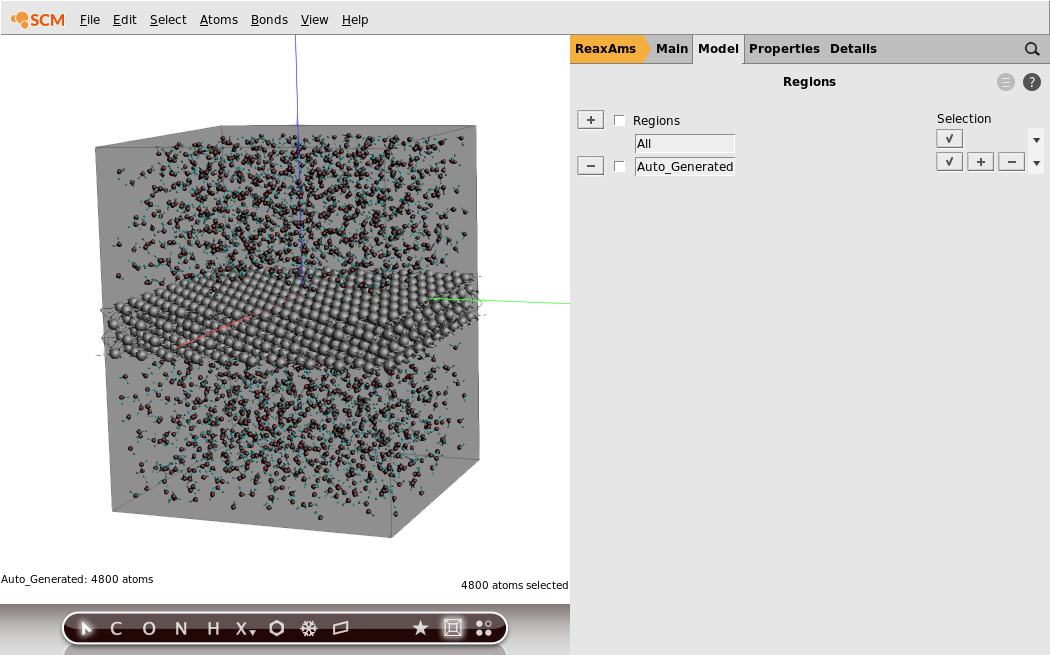
For clarity, let us rename the Auto_Generated region to Water:
- 1. Click and select the text
Auto_Generatedand change it intoWater
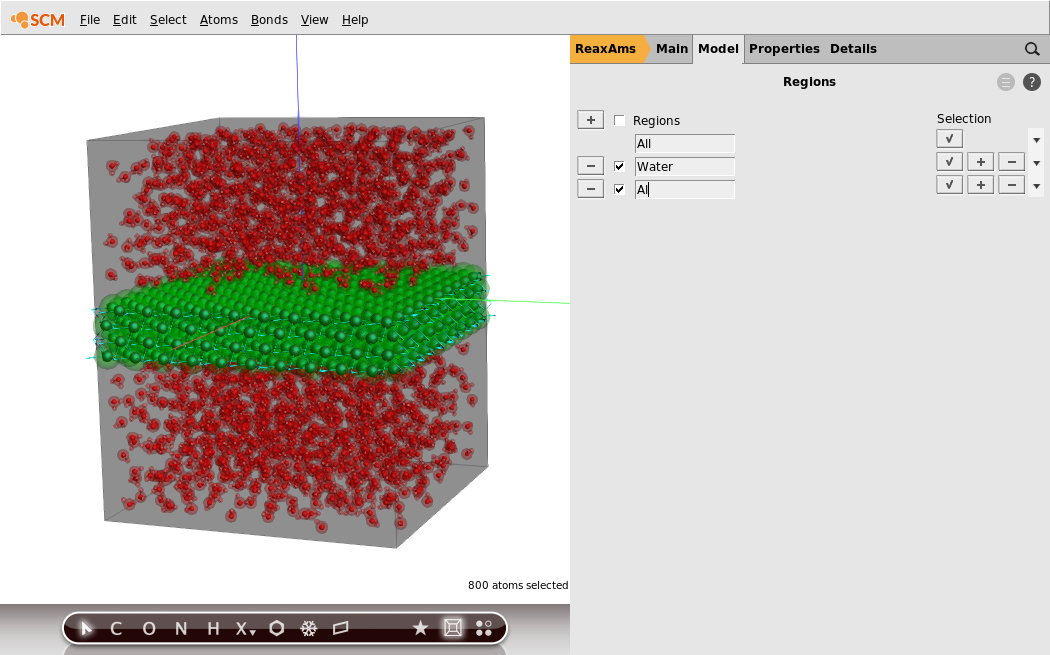
Now that we have a region defined that contains all water molecules, let us also make a region that contains just the aluminum slab.
- 1. Click on any of the aluminum atoms to select it.2. Select all other aluminum atoms by clicking
Select → Select atoms of the same type.3. Press the + button in front of the Regions label to add a new region (containing all the selected atoms)4. Change the region’s name fromRegion_2toAl5. Click the checkbox to the left of theWaterregion to highlight it
You should now see the two different regions highlighted in different colors:
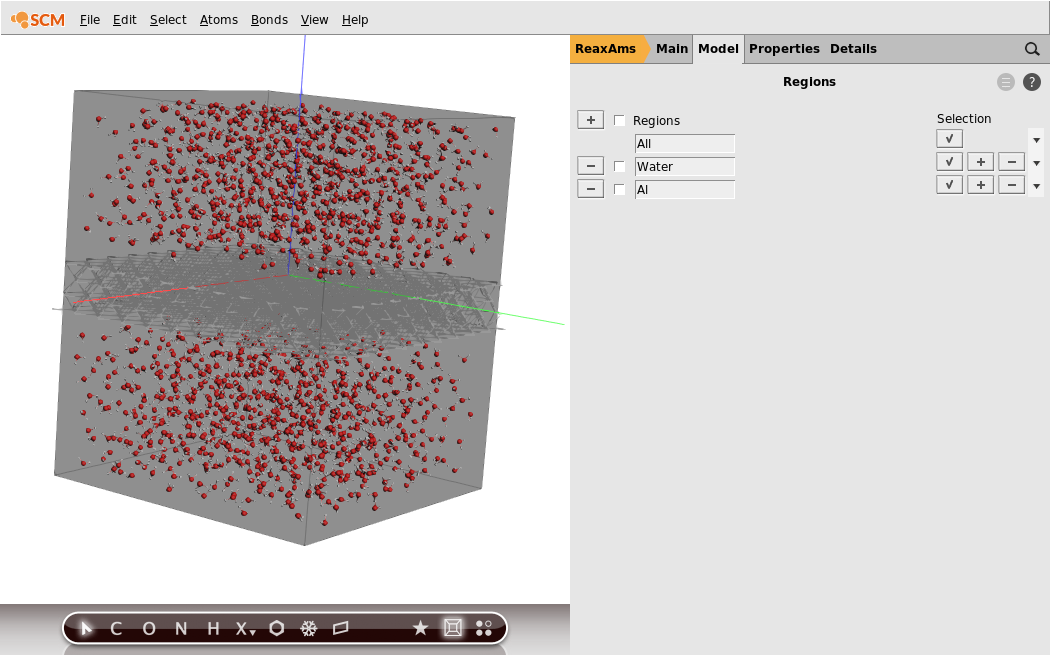
You can also set the visualization style per region:
- 1. Uncheck the check boxes at the left of the
WaterandAlregion lines2. Press the triangle on the right side of theAlregion line, and select the Wireframe visualization option
The aluminum slab is now shown in wireframe style.
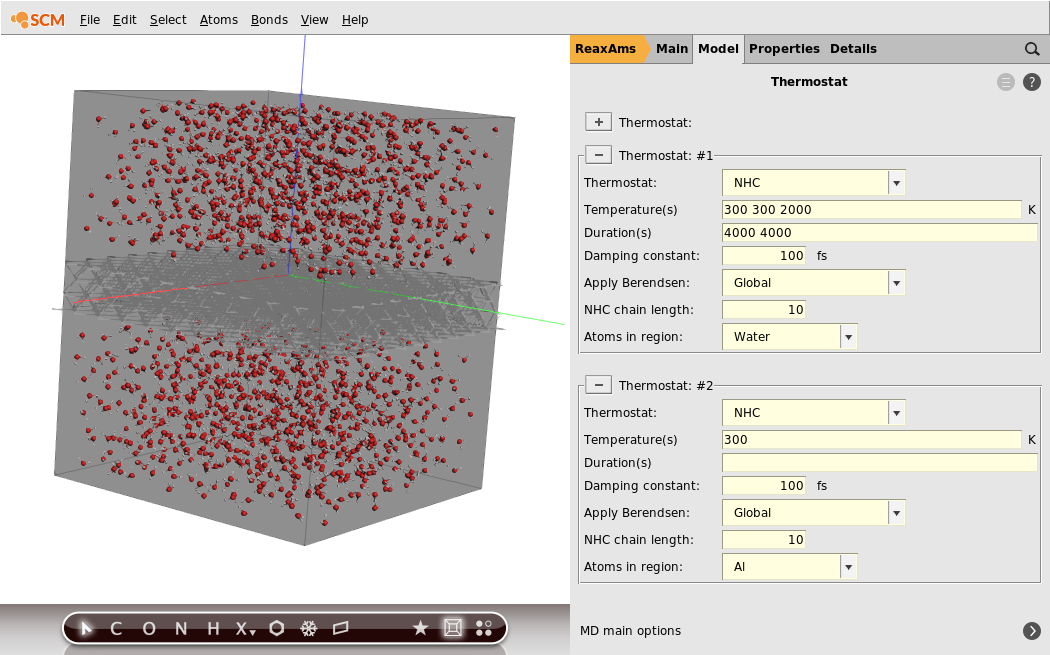
Now that we have defined the regions we needed, we will set up the temperature regime. We will start the water with a temperature of 300 K for 4000 steps, then warm it up to 2000 K within 4000 steps and maintain this temperature for the rest of the simulation.
- 1. Go back to the Model → Thermostat panel2. Select the Water region from the Atoms in region drop-down menu3. Type
300 300 2000into the Temperature(s) field4. Type4000 4000into the Duration(s) field
Hint
Note that with time-dependent thermostats the set temperatures are always connected with a linear ramp. The Temperatures field should therefore always contain one number more than the Durations field, as the last temperature will be held indefinitely. In the example above, in order to have a constant temperature of 300 K in the beginning, we explicitly specified that the temperature should go from 300 K to 300 K within the first 4000 steps.
We now have the time-dependent thermostat of the water set up. Let us add another thermostat that just keeps the aluminum slab cold.
- 1. Click the + button to create another thermostat3. Set the new Thermostat to NHC2. Set the new Temperature(s) to
300K4. Also set the Damping constant to100.0fs5. Select the Al region from the Atoms in region drop-down menu
Your thermostat setup should look like this:
Step 5: Run the simulation¶
Now we can run our set up:
- 1. File → Run2. When asked to save, specify
Al-wateras filename3. Follow your calculation by clicking SCM → Movie4. Rotate and zoom to get a good view of the surface5. Add a graph of the temperature by clicking Graph → Temperature6. Let the calculation run for a while (roughly until iteration 30000, that is frame 300)
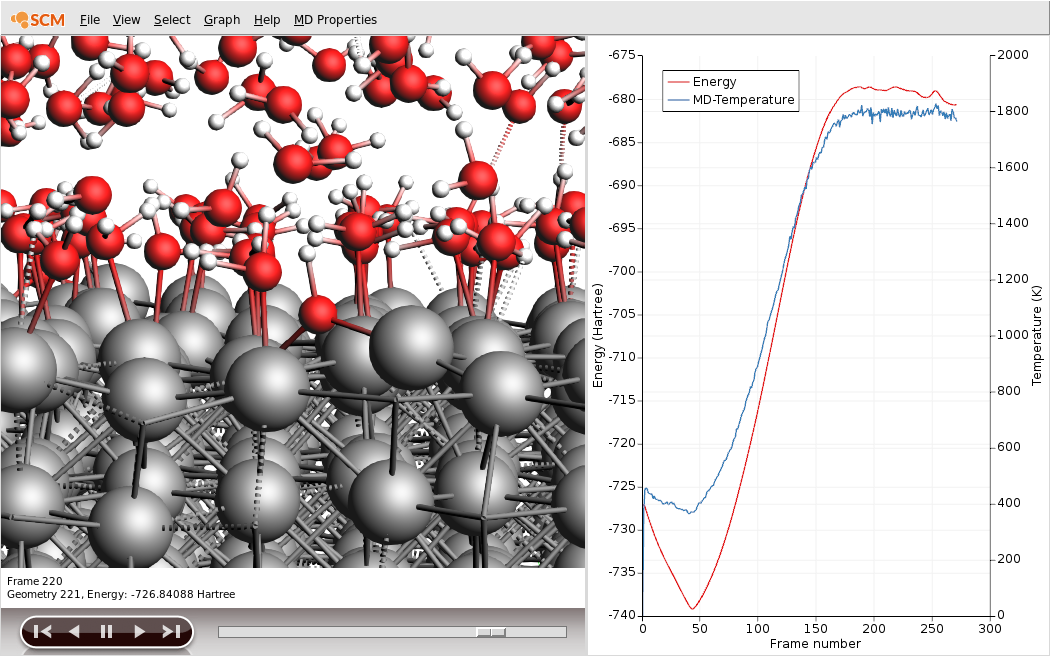
You will see many water molecules adsorb on the aluminum surface almost immediately. Around frame 40 (that is MD step 4000) you will see the temperature increase, as the water thermostat starts heating its region. After some time, you will also see some of the adsorbed water molecules loose one hydrogen. The lone proton will likely drift off into the water, while the remaining OH goes into a bridge-like configuration with two of the surface Al. This configuration can already be seen in the picture above.
You can leave the simulation running to see what will happen. It will take a long time though. If you do leave it running for a while, you will see that the OH in the aforementioned bridge configuration tend to pull one of the Al atoms out of the surface, essentially roughening it and allowing more water to adsorb. You might also see aluminum atoms completely detach from the surface an become dissolved in the water layer as AlO3H3. Eventually a rough aluminum oxide layer forms, while the split off protons combine to form molecular hydrogen.
You can download the movie here if it does not play in your browser.
We can use the molecular composition analysis tools in ADFmovie to get a bit clearer picture of that process. Let us plot the number of H2O and H2 molecules in our simulation and also hide all the water molecules in the movie.
- 1. Click Graph → Add Graph2. Click MD Properties → Molecules3. Tick the Graph check-boxes for H2O and H24. Untick the Show check-box for H2O to hide all the water molecules
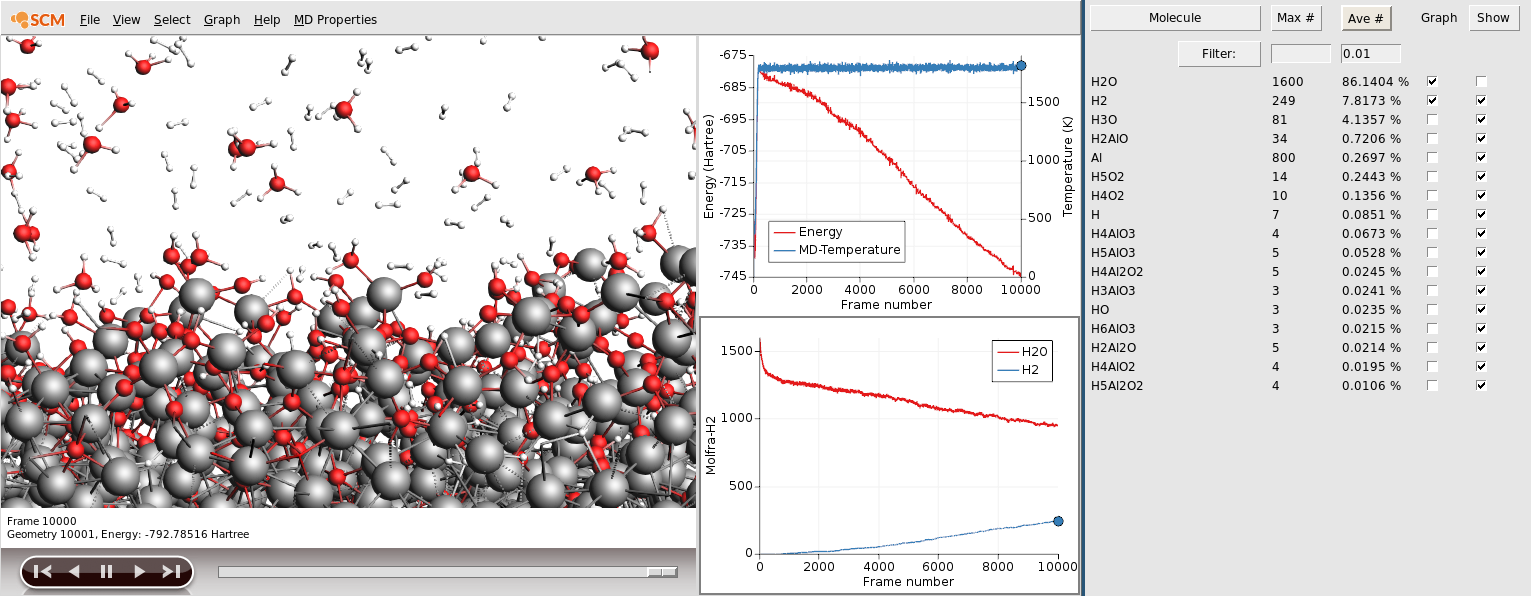
However, you will likely need to let your calculation run over night to see this happening. If you do not want to wait for the simulation to finish, you can now kill the job:
- 1. Bring the ADFjobs window to the front2. Make sure your Al-water job is selected (click once on it if not)3. Kill it using Job → Kill4. To close all GUI modules click SCM → Quit All