Toluene charges, movies, frequencies and normal modes¶
The MOPAC-GUI is basically the same set of programs as the ADF-GUI. We suggest you start with the more general GUI tutorials if you are not familiar with the ADF-GUI.
Set up Toluene in MOPAC¶
- Start ADFjobsSCM → New InputSwitch to MOPAC:
 →
→ 
We will now make a Toluene molecule. Most conveniently this is done by simply searching for it:
- Control/Command-F or click on
 Enter ‘toluene’ as search textSelect ‘Toluen’ (the second match) from the list of matchesClick once in the drawing area in empty space to clear the selection
Enter ‘toluene’ as search textSelect ‘Toluen’ (the second match) from the list of matchesClick once in the drawing area in empty space to clear the selection

Run interactively¶
In the Mopac Main panel you can see what options for MOPAC have been selected: by default a Geometry optimization will be performed, using the PM7 method. MOPAC will also calculate the bond orders, and the GUI will automatically update the molecular bonds using these calculated bond orders. To show this, we will first remove the bonds and then run MOPAC by pressing the Run button:
- Bonds → Remove BondsClick on the ‘Pre-optimize’ button in the lower right corner
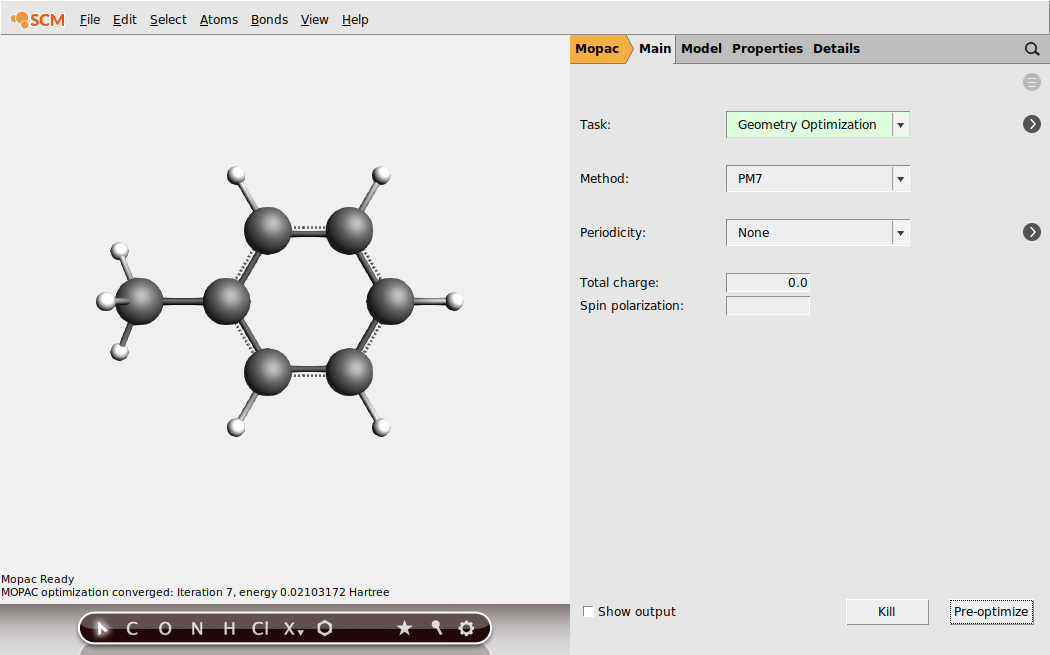
The geometry has been optimized, but as the starting geometry was very reasonable this did not lead to big changes. In the status area you can see the status of the MOPAC pre-optimization and the final heat of formation. And obviously the bonds reappeared. These bonds are actually based on the bond orders as calculated by MOPAC.
You can now also show the calculated atomic charges:
- View → Atom Info → Atomic: Charge (mopac) → Show
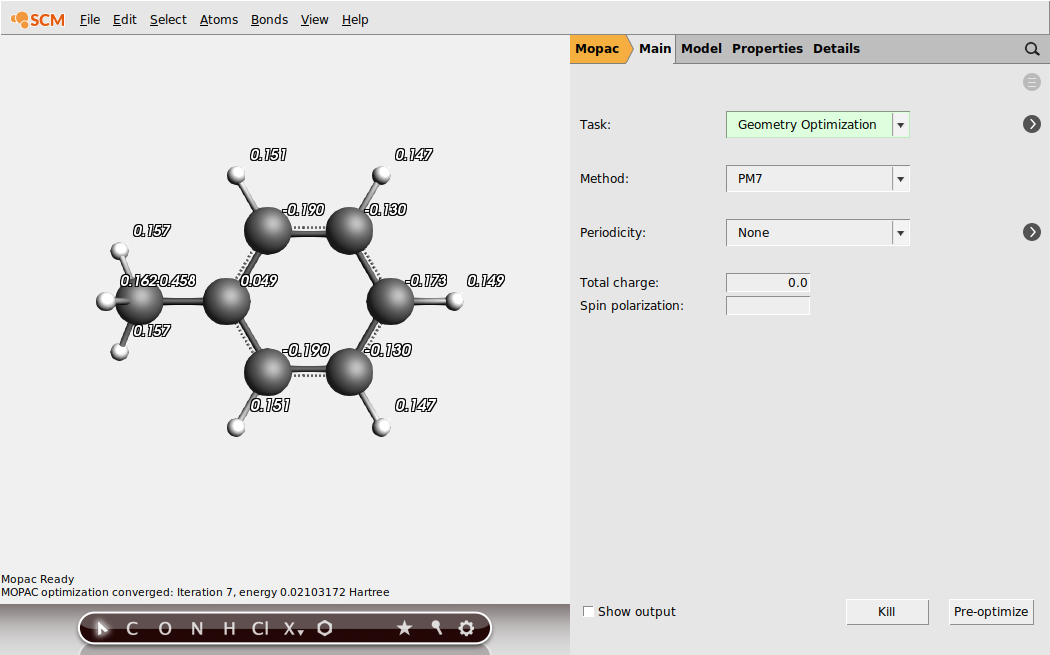
- Close all windows except ADFjobs (no need to save your setup)
Save job and results: movies, IR spectrum and normal modes¶
The previous section showed how to run MOPAC interactively, typically to be used as a pre-optimizer. However, you can also run the MOPAC calculation as a ‘regular’ job. Then the result files will be saved, and you can analyze them with the GUI tools.
- SCM → New InputPanel bar ADF → MopacControl/Command-F or clicl onEnter ‘toluene’ as search textSelect ‘Toluen’ (the second match) from the list of matchesClick once in the drawing area in empty space to clear the selection
Once again we have our Toluene molecule. We now request the calculation IR spectrum and normal modes:
- In the top panel, click on Properties → Frequencies, Photons, Elastic Tensor and check the Frequencies checkbox
Now to run it as a job simply use the Save and Run command from the menu:
- File → Save, save the job with name ‘Toluene’File → Run
- When asked to read the new coordinates (when the calculation finishes), click YesSCM → Logfile
The logfile will show the progress of the calculation:

- SCM → ViewProperties → Atom Info → Atomic: Charge (mopac) → Show

Now for a geometry optimization it might be interesting to see a movie of the progress of the optimization:
- Close the ADFoutput windowSCM → MovieGraph → Energy

You can also use ADFmovie to get detailed information about the geometry (distances, angles and so on), just as for an ADF geometry optimization. If you don’t know how, check the more general GUI tutorials.
The calculated IR spectrum can be visualized via ADFSpectra:
- SCM → Spectra
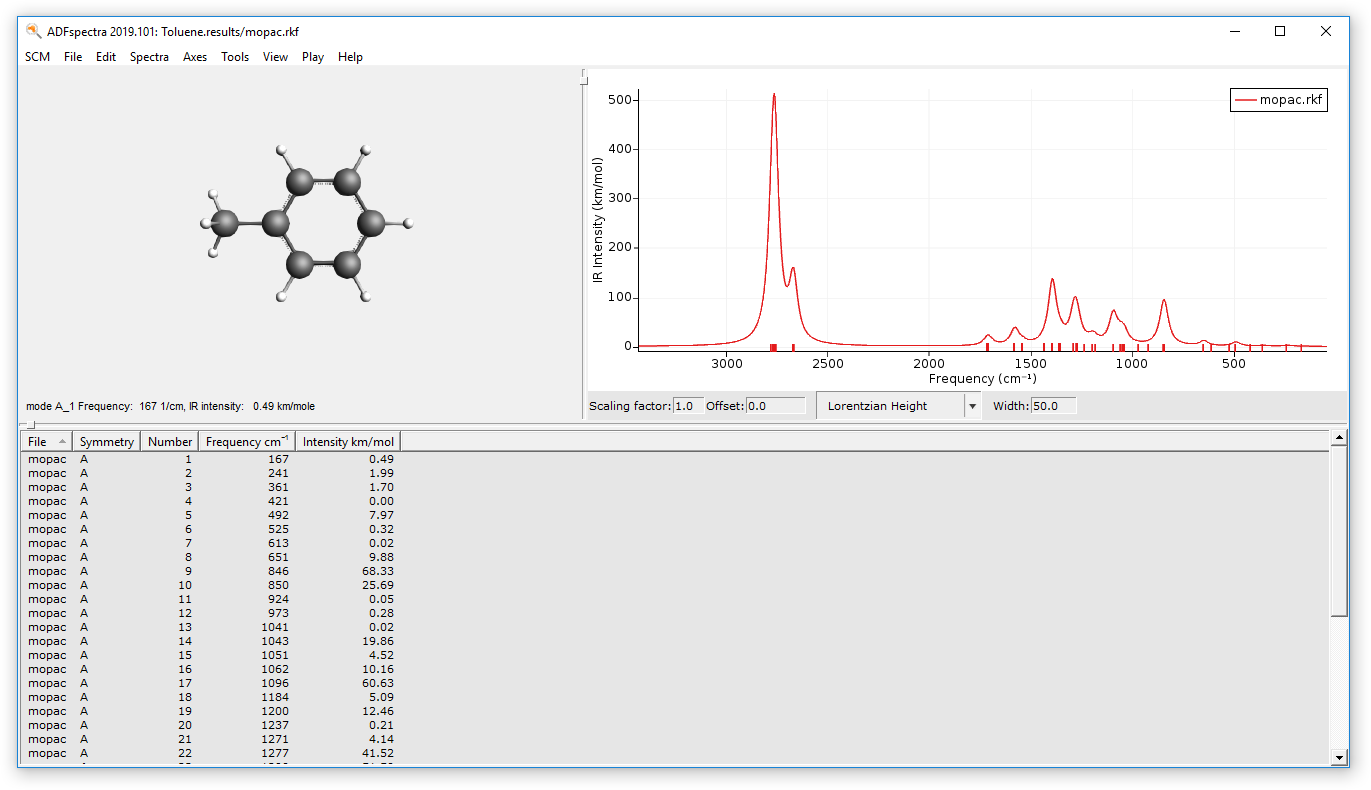
- Click on the peak with the highest intensity, which is found around 2700-2800 cm-1.
The selected mode will be shown:
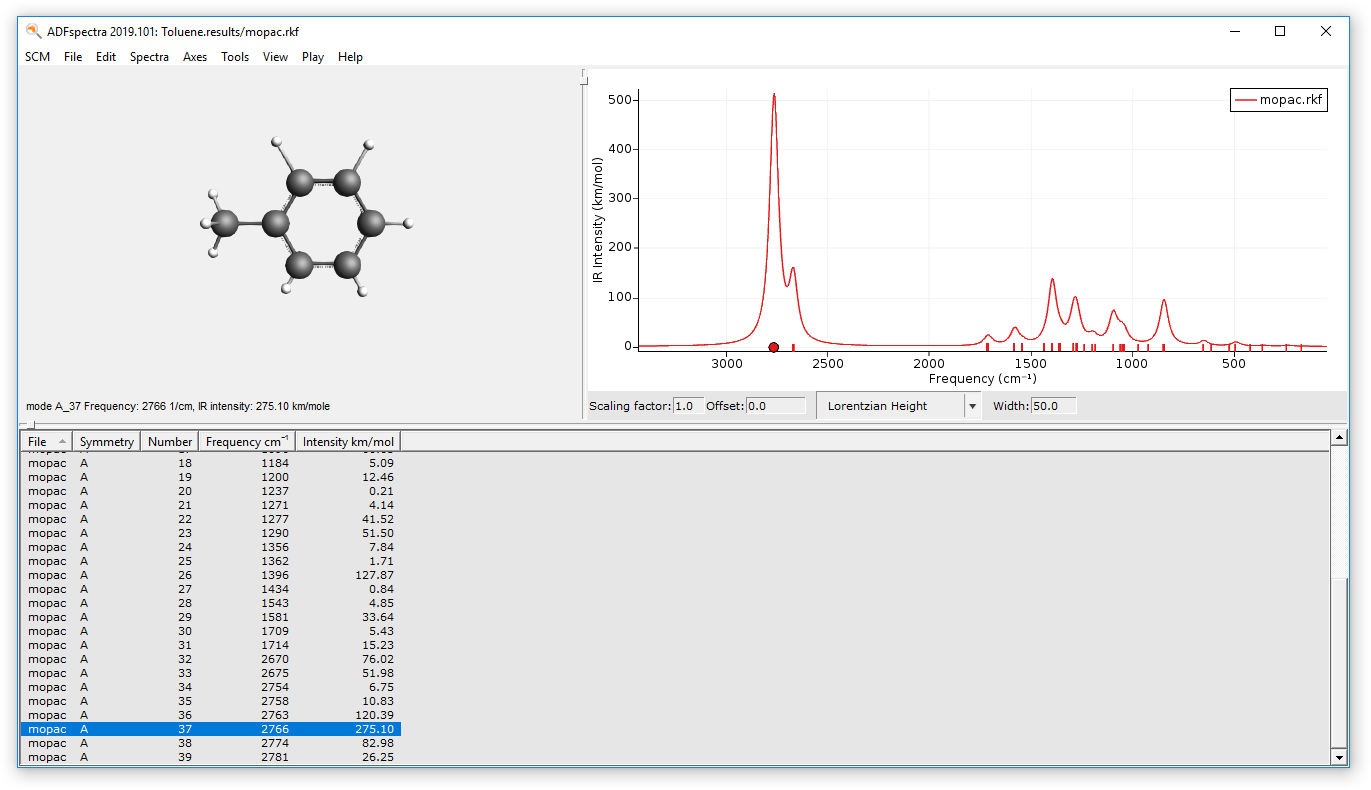
- Browse through the normal modes using the left and right arrow keyTry the alternative method:Click on the normal mode of interest in the tableSCM → Quit All