Getting started with BAND¶
BAND is an atomic-orbital based DFT program for periodic systems (crystals, slabs, chains and molecules).
This introductory tutorial will show you how to:
- Set up and run a BAND calculation (using ADFJobs and ADFInput)
- Visualize the band structure and density of states using BandStructure
- Visualize densities and atomic charges with ADFView
If you are not at all familiar with our Graphical User Interface (GUI), check out the Introductory tutorial first.
Create a work directory and start up ADFInput¶
Let us begin by creating a new work directory for our tutorial using ADFjobs:
- Start ADFjobsSelect File → New DirectoryRename the new directory by typing, for example, ‘BandTutorial’ and hitting ReturnMove into that directory by clicking on the corresponding folder icon
Now we can start ADFInput from the SCM menu:
- Click on SCM → New Input
In ADFInput switch the program from ADF (our molecular DFT program) to BAND:
- Switch to BAND:
 →
→ 
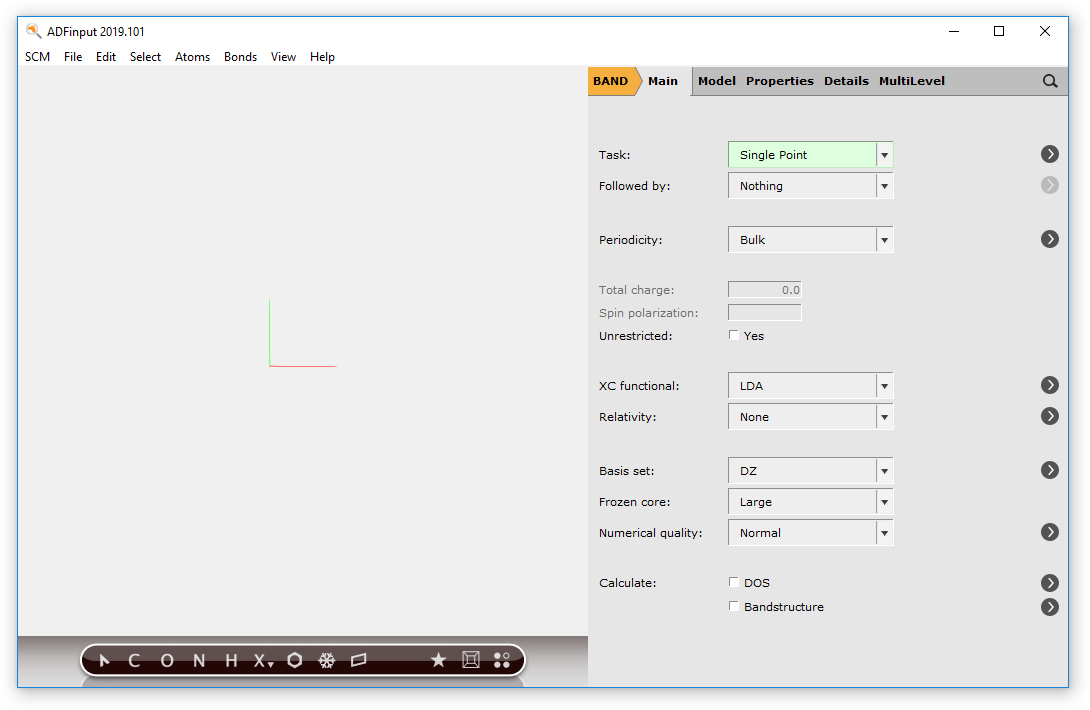
Set up the NaCl crystal calculation¶
We now want to create a sodium chloride crystal. Let us import the geometry from our database of structures:
- 1. search for NaCl in the search box
 2. select Crystals → NaCl3. click on View → Periodic → Repeat Unit Cells
2. select Crystals → NaCl3. click on View → Periodic → Repeat Unit Cells

There are other ways of defining the geometry of your system:
- You could draw your system in the molecule editing area, as described in the tutorial Building Crystals and Slabs.
- You could import the geometry from a file: File → Import Coordinates… (
Download NaCl.xyz). Supported formats: .xyz, .cif and .pdb.
We will now set up the calculation details in the Main Panel: in this tutorial we will perform a single point calculation with the PBE XC functional, and we will compute the Band Structure and Density Of States (DOS).
- 1. On the Main panel, click on the XC functional drop-down menu and select GGA → PBE2. Tick the DOS and Band structure check boxes

Run the calculation¶
First we need to save our job:
- Click on File → Save
and then we can run the calculation:
- Click on File → Run
This will open ADFjobs and start the calculation

You can monitor the progress of your calculation by opening the log file:
- Select your job in ADFJobsclick on SCM → logfile

Once the calculation is finished, we can visualize the results.
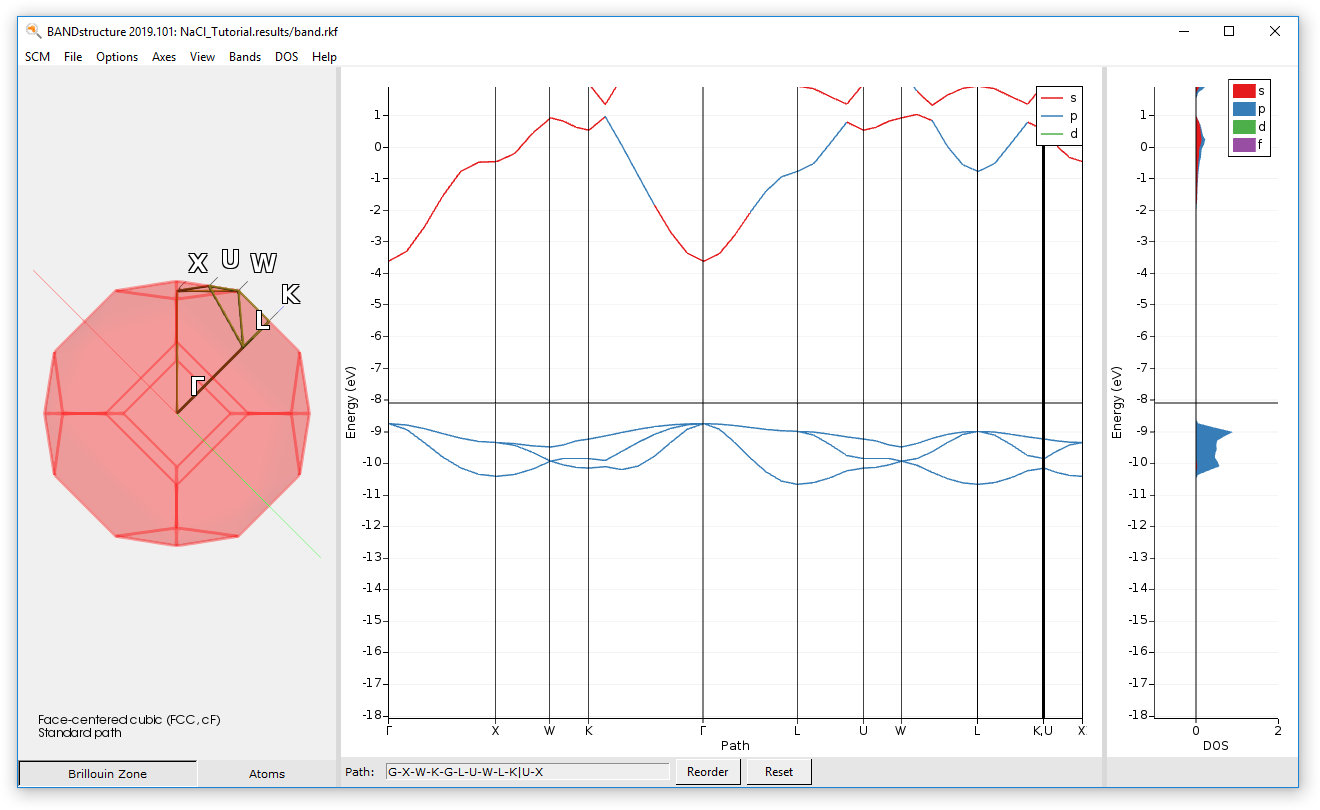
Examine the band structure and DOS¶
With the BandStructure program we can inspect the band structure and density of states. To open BandStructure:
- Select the job in ADFJobsclick on SCM → Band Structure
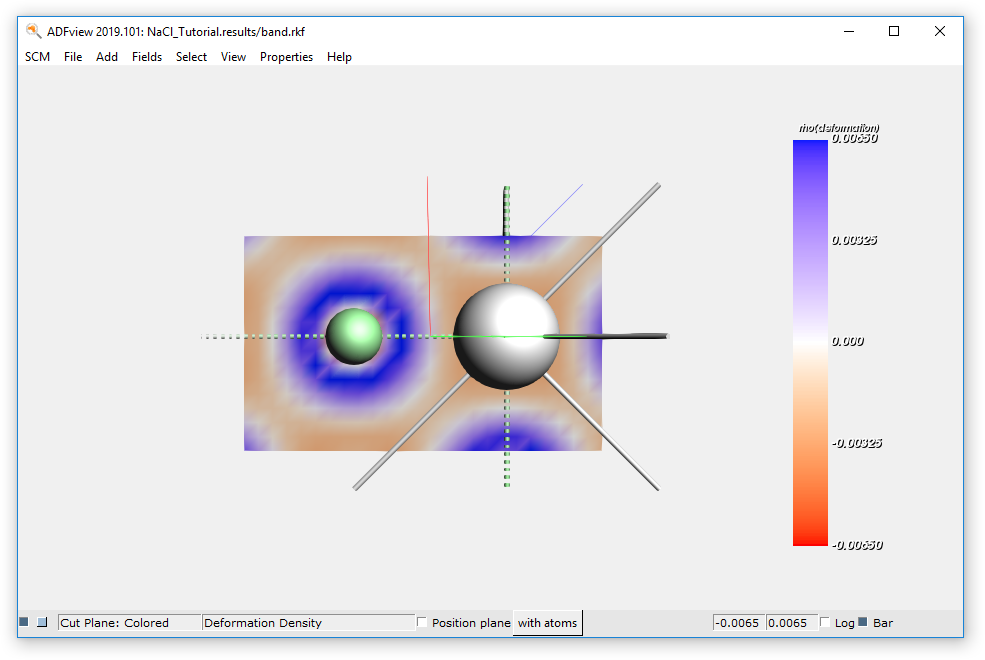
Visualize results with ADFView¶
Three-dimensional fields can be visualized with ADFView
- Select your job in ADFJobsclick on SCM → View
See also
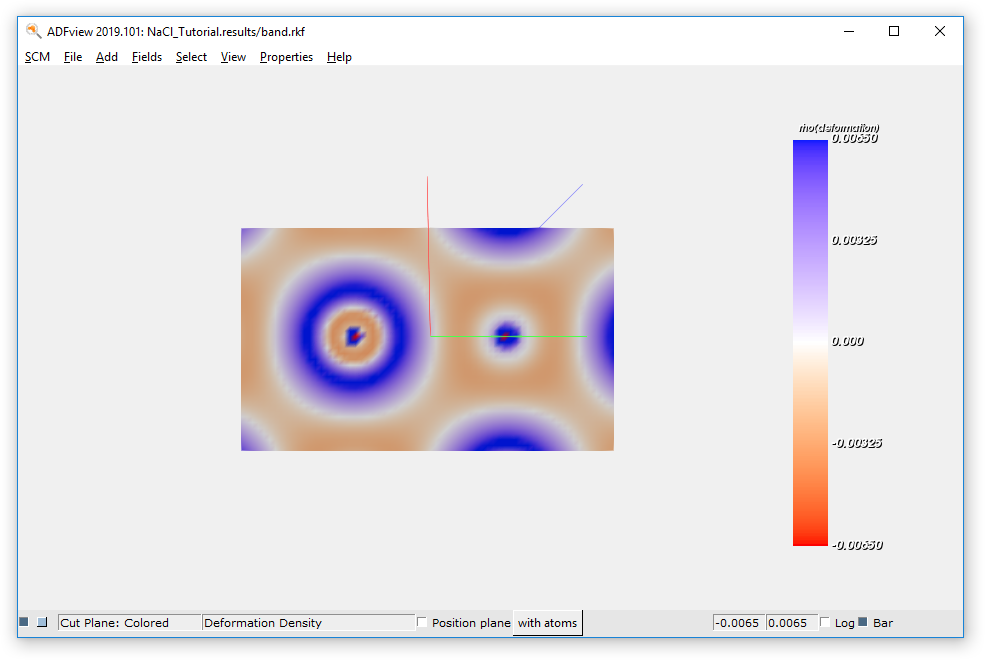
Let’s visualize the deformation density (i.e. the difference between the electronic density in the crystal and the electronic density of the spherical spin-restricted isolated atoms) on a cut plane:
- Click on Add → Cut Plane: ColoredClick on Select Field → Density → Deformation DensityTweak the various parameters, as showed in the following picture
Tip
If you hover with the mouse cursor over the min and max fields values, you can see the actual minimum and maximum values of the field.

To hide the atoms:
- Click on View → Molecule → Hidden
To increase the resolution of our cut-plane plot:
- Click on Fields → Grid → Medium
This concludes the Getting Started tutorial.