Basis set superposition error (BSSE)¶
This tutorial will show you how to do calculate a basis set superposition error (BSSE) using ADF for a formamide dimer. The first method uses atomic fragments, the second method uses molecular fragments.
In this example double-hybrids are used. Note that this example is not a typical example in which double-hybrids improve much over dispersion-corrected hybrid functionals. However, this example is relevant for double hybrids, since the BSSE is typically larger for double-hybrids than for standard GGA or hybrid functionals. Thus for double-hybrids it is especially relevant to correct for a BSSE.
An all electron TZ2P basis set is used since that is the recommended choice in ADF for double hybrids. For reasons of numerical stability, it is also highly recommended not to use larger or more diffuse basis sets. The use of triple-zeta quality basis sets is usually necessary for accurate double-hybrid energies.
Method 1: atomic fragments¶
- Start a new ADFinputCopy-paste the following coordinates of a formamide dimer:
C 2.01911735 -0.03015376 0.00000000
C -2.01911735 0.03015376 0.00000000
N 1.39482051 1.15825415 0.00000000
N -1.39482051 -1.15825415 0.00000000
H 0.37362012 1.21206244 0.00000000
H 3.11670830 0.04878624 0.00000000
H 1.94222103 1.99902922 0.00000000
H -1.94222103 -1.99902922 0.00000000
H -0.37362012 -1.21206244 0.00000000
H -3.11670830 -0.04878624 0.00000000
O 1.46498124 -1.12721089 0.00000000
O -1.46498124 1.12721089 0.00000000
- XC functional: Double Hybrids → B2PLYP-D3BJ.Basis set: TZ2P.Frozen core: None.Click on File → Save As… and give it the name “Formamide_dimer”.Click on File → Run.Wait for the calculation to finish.
- Select all atoms of one of the formamide monomers (holding shift)Select Atoms → Ghosts → Change Atoms to GhostsClick on File → Save As… and give it the name “Formamide_BSSE”.Click on File → Run.Wait for the calculation to finish.

Note that the geometry of the two monomers is exactly the same apart from a rotranslation. The BSSE corrected energy of the Formamide dimer with respect to its monomer fragments can now be calculated as the bond energy printed in Formamide_dimer.logfile minus two times the bond energy printed in Formamide_BSSE.logfile, which is approximately -15.6 kcal/mol.
Method 2: molecular fragments¶
- Start a new ADFinputCopy-paste the coordinates of the formamide dimer of method 1XC functional: Double Hybrids → B2PLYP-D3BJ.Basis set: TZ2P.Frozen core: None.Panel bar Model → RegionsSelect all atoms of one of the formamide monomers (holding shift)Click the ‘+’ button to add a new region (containing the atoms of 1 monomer)Change the name of ‘Region_1’ into ‘monomer’Select Atoms → Invert SelectionSelect Atoms → Ghosts → Change Atoms to Ghosts

- Panel bar MultiLevel → FragmentsCheck the ‘Use fragments’ check boxClick on File → Save As… and give it the name “Formamide”.Click on File → Run.Wait for the 2 calculations to finish.
The BSSE for the formamide monomer which is the energy of the monomer calculated in the dimer basis set minus the the energy of the monomer calculated in its own monomer basis set is printed as bond energy in Formamide.logfile, which is approximately -0.88 kcal/mol.
Next we calculate the formamide dimer using the formamide monomer as fragment. Note that the geometry of the two monomers is exactly the same apart from a rotranslation, which means that the same monomer fragment can be used.
- Start a new ADFinputCopy-paste the coordinates of the formamide dimer of method 1XC functional: Double Hybrids → B2PLYP-D3BJ.Basis set: TZ2P.Frozen core: None.Panel bar Model → RegionsSelect all atoms of one of the formamide monomers (holding shift)Click the ‘+’ button to add a new region (containing the atoms of 1 monomer)Select Atoms → Invert SelectionClick the ‘+’ button to add a new region (containing the atoms of the other monomer)Panel bar MultiLevel → FragmentsCheck the ‘Use fragments’ check boxClick on the folder next to Region_1: and select Formamide.monomer.t21Click on the folder next to Region_2: and select Formamide.monomer.t21
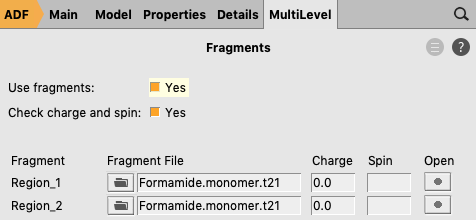
- Click on File → Save As… and give it the name “Formamide_dimer_EDA”.Click on File → Run.Wait for the calculation to finish.
The uncorrected energy of the Formamide dimer with respect to its monomer fragments can now be read from the Formamide_dimer_EDA.logfile and is approximately -17.39 kcal/mol. After correction this energy for BSSE this makes -17.39 - 2*(-0.88) = -15.6 kcal/mol as was found in method 1.
Note that in the energy decomposition analysis (EDA) in case of double-hybrids is incomplete. The bonding energy with respect to the individual fragments is calculated and printed out. However, all terms which rely on the orthogonal fragment density (e.g. Pauli repulsion terms) do not include the MP2 part of the double-hybrid.