Using the UNIFAC program¶
The UNIFAC program is an implementation of the UNIFAC method, a fast and accurate liquid-phase activity coefficient model. One of the main advantages of the UNIFAC method is that, unlike COSMO-RS/-SAC methods, it does not require any ADF calculations and can estimate thermodynamic properties based only on SMILES strings representations of every molecule in the system. This means that the UNIFAC method can be applied to liquid systems for which no 3D/surface charge information is known. Additionally, UNIFAC is very efficient, able to generate thermodynamic property estimates in milliseconds. In the following sections, we review basic GUI functionality for the UNIFAC program.
Selecting/inputting compounds¶
Molecules can be used with the UNIFAC program in two ways:
(1) Simply chosing them from the ADFCRS compound database.
(2) Inputting a SMILES string in Compounds → Add Compound using QSPR (Fast Sigma). → Add. This will generate a .compkf file, which will appear in the compound database and can be used with UNIFAC calculations.
Caution
In the current version of the GUI, you can only add a compound by SMILES string if the method COSMO-RS or COSMO-SAC is selected. It will not work if UNIFAC is selected.
To input a molecule as a SMILES string, simply follow the instructions above, typing the SMILES string in the field. As an example, we input cyclohexanol:
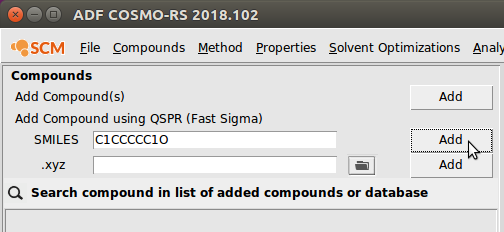
This will prompt you to save your compound as a .compkf file. After saving it, it should appear in the database.
Inputting property values¶
For some types of UNIFAC calculations, physical property data will be required. For most applications, all of the required physical property data can be input from the compounds menu: Compounds → List of Added Compounds. Property input fields are given in the right panel. As an example, we input antoine coefficients for cyclohexanol:

Note that other properties (enthalpy of fusion, melting point, etc.) can be input or estimated with a Property Prediction Method.
Calculations with the UNIFAC program¶
To begin, first navigate to the UNIFAC method:
- Start ADFcrs (if not already open)Select Method → UNIFAC
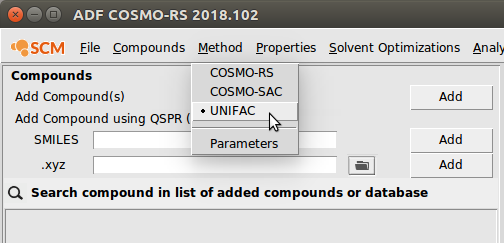
All of the available properties for UNIFAC are listed under Properties →:

Pure Compound Properties provides estimated physical properties for a compound, but the rest require a UNIFAC calculation. The rest are summarized in the following subsections.
Vapor Pressure Mixture¶
- Properties → Vapor Pressure Mixture
For this calculation, we also select water from the ADFCRS database. We input the following antoine parameters for Water: 4.56, 1435.26, -64.85. Next, we navigate to the Vapor Pressure Mixture window. We choose a two component mixture of 0.8:0.2 cyclohexanol:water and change the temperature range to 250-350 K. This looks like the following:

Running this calculation produces a graph display total and partial pressures of the mixture over the temperature range.
Activity Coefficients¶
- Properties → Activity Coefficients
Next, we demonstrate the activity coefficient calculation module. In this example calculation, we calculate the activities of a Methanol/Water system (both compounds were selected from the ADFCRS database). Additionally, we calculate the activity coefficient of aniline at infinite dilution in this system:

Partition Coefficients (LogP)¶
- Properties → Partition Coefficients (LogP)
In this example, we calculate the logP of Ibuprofen, Thymol, and Xylitol in the standard Octanol/Water system.

Caution
Only the presets work in the current implementation. User-defined solvent systems will not work.
Solubility in Pure Solvents¶
- Properties → Solubility in Pure Solvents
In this example, we calculate the solubility of Ibuprofen in a few different solvents: Methanol, Water, and Cyclohexanol (from our input SMILES string). We also want to consider the solibility over the temperature range 250-198.15 K. Because Ibuprofen is a solid at these temperatures, we require its Enthalpy of Fusion and Melting Point. We can either enter experimental values or estimate the values in the Compounds → List of Added Compounds menu. Even though these properties are known experimentally, we will estimate them as shown below:

Now, we can navigate to Properties → Solubility in Pure Solvents and input the following:

Solubility in Mixture¶
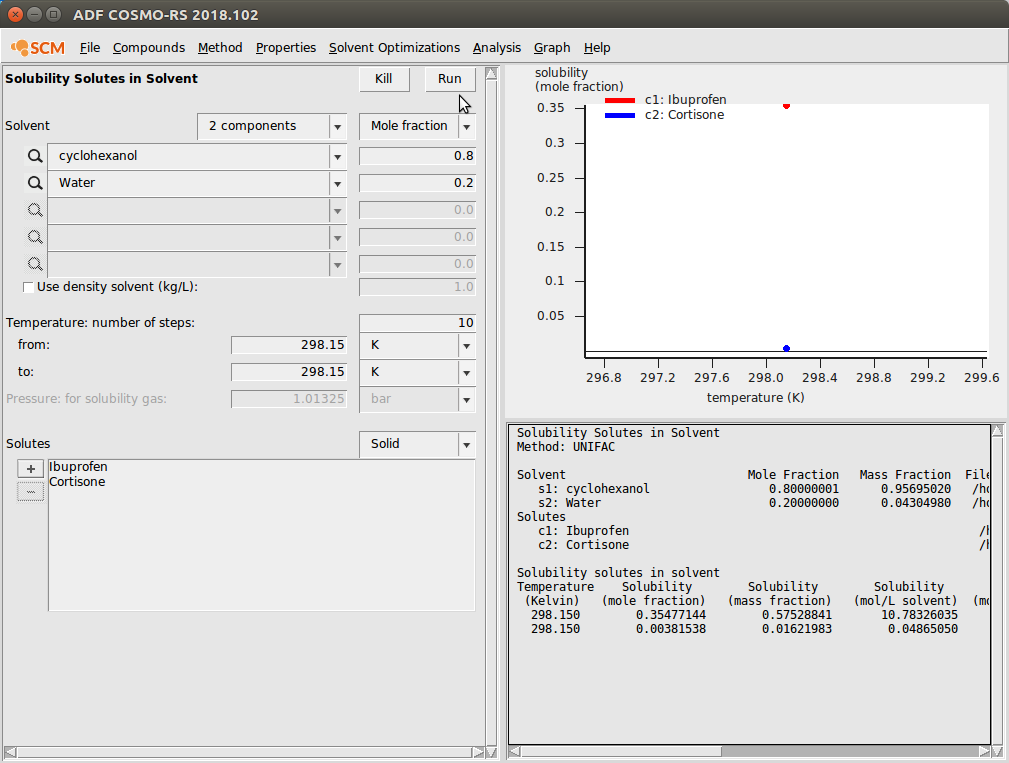
- Properties → Solubility in Mixture
Solubility in Mixture is very similar to Solubility in Pure Solvents. In this case, we specify a mixture of cyclohexanol and water and estimate the solubilities of Ibuprofen and Cortisone in that mixture.
Binary Mixture VLE/LLE¶
- Properties → Binary Mixture VLE/LLE
To demonstrate the Binary Mixture module, we again use our cyclohexanol/water system because we’ve already input the antoine coefficients. If no antoine coefficients are found, the vapor pressure will be set to 0 and VLE data will not be meaningful. This calculation looks like the following:
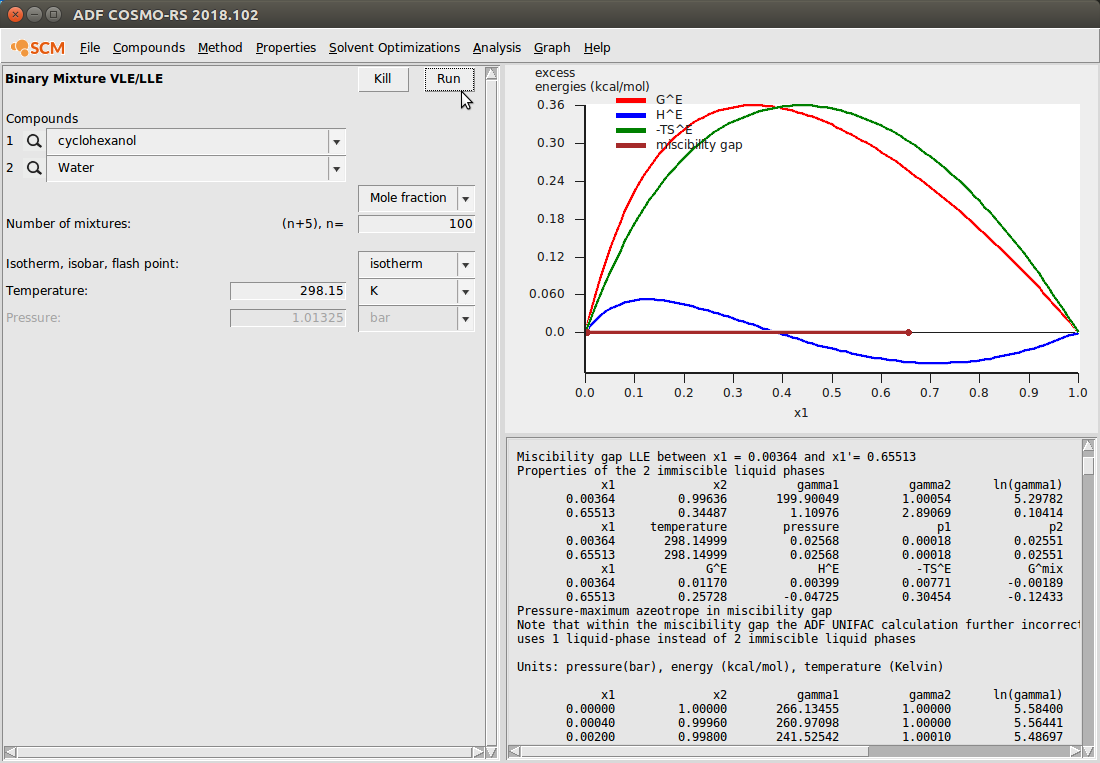
Note that here we’ve changed the default graph display to view the excess energies. This can be done with Graph → Y Axes → excess energies.
Ternary Mixture VLE/LLE¶
- Properties → Ternary Mixture VLE/LLE
Next, we add a diethyl ether to our cyclohexanol/water system to investigate the behavior of the ternary mixture. We input the following Antoine coefficients for Diethyl ether (Ethoxyethane): 4.022, 1062.64, -44.93. The calculation produces the following:
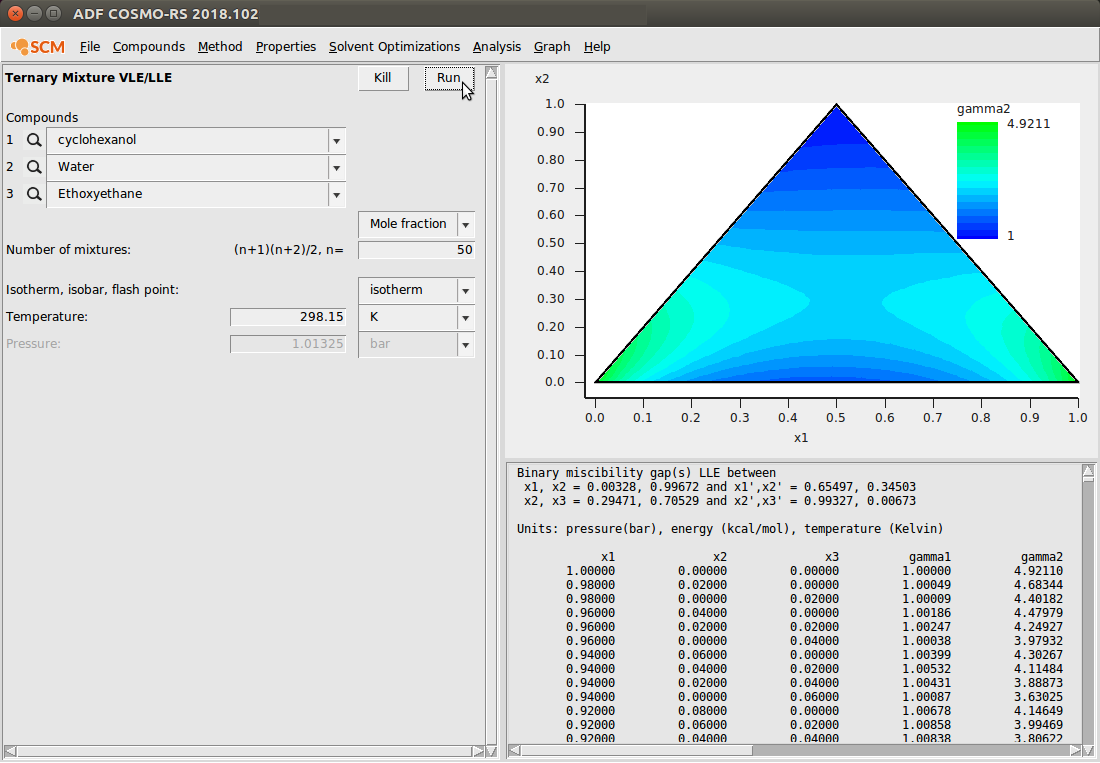
Also note that in this example, we’ve changed the default graph display to a ternary plot with Graph → Triangle \(\boldsymbol \Delta\). Also, the background color has been changed to show the activity coefficient of compound 2 (water) at the different mole fractions. This was done with Graph → Z Colormap → gamma2.
Common issues¶
The most frequently encountered issue with the UNIFAC program is termination due to lack of binary interaction parameters. Unlike COSMO-RS/-SAC, UNIFAC requires binary interaction parameters for every pair of molecular substructures – called groups – in the mixture. There are many cases where these binary interaction parameters do not exist or are not publicly known. For molecular systems with even one missing interaction parameter, no UNIFAC estimate can be provided. At present, our group coefficients and interaction parameters come from The Dortmund Data Bank UNIFAC site.