Training sets for ReaxFF Reparametrization¶
Co.ff¶
The cobalt training set documented here was used in the parametrization of the Cobalt force field, Co.ff, published in “Development of a Transferable Reactive Force Field for Cobalt” by M. R. LaBrosse et al. Within the publication the choice of training set entries is discussed from a scientific standpoint. The purpose of this page is to demonstrate how the data translates into the format of the trainset.in and geo files used by the force field optimizers shipped with the ADF modeling suite.
Note
This single-element training set does not contain any charges. Further note that the default run type (RUTYPE NORMAL RUN in the geo file) is set to a geometry optimization in the ReaxFF control file, so unless noted otherwise, every training set geometry is optimized (fixed lattice) by ReaxFF every time the objective function is evaluated.
Contents:
- Weighting of individual entries
- General energies, cluster models and the Co2 dimer
- Crystaline phases
- General Co surfaces
- Adatoms
- Vacancies and defects
- Elastic strain moduli
The complete training set (trainset.in, geo, control) can be requested from here . Documentation regarding the format of the input files for the optimizers can be found here here .
Weighting of individual entries¶
To estimate the quality of a given set of ReaxFF parameters with respect to the trainingdata an objective or error function is defined:
where the sum runs over all training set entries. Each difference between the reference property (xi,TS) and the ReaxFF value (xi,ReaxFF) is weighted individually via the weightings σi.
Note
The value of this objective function is the quantity that is minimized by the optimization algorithms used for the force field fitting. It is therefore important to choose the weightings such that there is no unwanted bias towards one or the other entry.
In total the Co training set contains as little as 144 entries all of which are energies defined inside a single ENERGY block of the trainset.in file:
ENERGY
[...]
ENDENERGY
Inside the energy block the training data is distributed as follows:
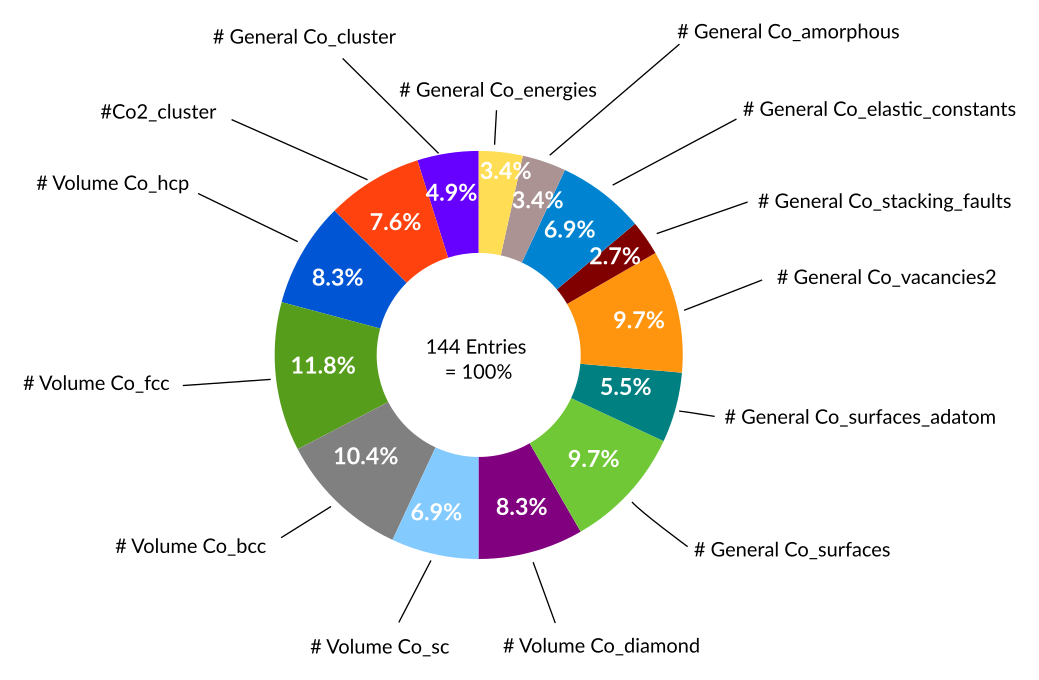
The contribution of each entry to the overall error function can to some extend be visualized using the weightings, σi, of the error function.

For example, the most stable phases hcp and fcc are given much higher weights during the optimization (25.4% and 36.0%) than the less favourable diamond phase (0.5%). In practice one would use the breakdown of the error function provided by the optimizers (fort.99 file) to finetune the weights of the objective function, which is probably how the above weightings were set too.
General energies, Cluster models and the Co2 dimer¶
The energy differences between optimized cubic phases are included in the training data
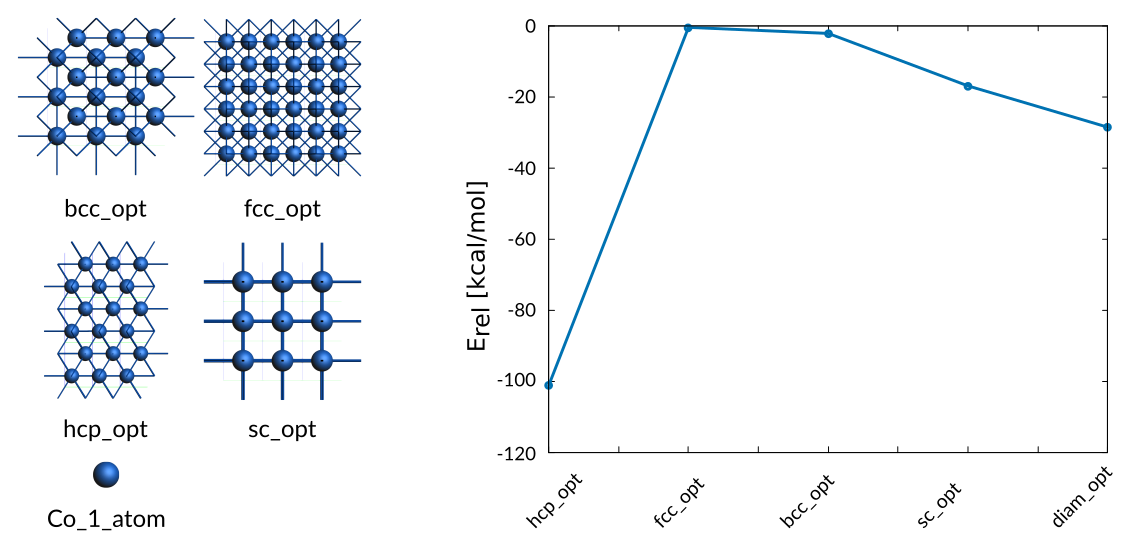
the according entry in the trainset.in file is found towards the end:
# General Co_energies
0.50 + hcp_opt/2 - Co_1_atom/1 -101.30
0.02 + hcp_opt/2 - fcc_opt/4 -0.52
0.10 + hcp_opt/2 - bcc_opt/2 -2.21
1.00 + hcp_opt/2 - sc_opt/1 -17.00
2.00 + hcp_opt/2 - diam_opt/8 -28.60
In addition cohesive energies for a set of small clusters of sizes 2, 3, 4, 5, 6, 8, and 13 atoms are included

cohesive energies entry in trainset.in:
# General Co_clusters
2.00 + Co_2_atom/2 - Co_1_atom/1 -29.79
2.00 + Co_3_atom/3 - Co_1_atom/1 -38.80
2.00 + Co_4_atom/4 - Co_1_atom/1 -46.94
2.00 + Co_5_atom/5 - Co_1_atom/1 -56.91
2.00 + Co_6_atom/6 - Co_1_atom/1 -64.58
2.00 + Co_8_atom/8 - Co_1_atom/1 -65.68
2.00 + Co_13_atom/13 - Co_1_atom/1 -71.06
A scan of the bond stretch for the Co2 dimer is included as well

scan for the Co2 dimer in trainset.in:
# General Co2_cluster
5.00 + dimer_sep_1/2 - Co_1_atom/1 -22.75
5.00 + dimer_sep_2/2 - Co_1_atom/1 -27.36
5.00 + dimer_sep_3/2 - Co_1_atom/1 -29.43
5.00 + dimer_sep_4/2 - Co_1_atom/1 -29.79
5.00 + dimer_sep_5/2 - Co_1_atom/1 -29.75
5.00 + dimer_sep_6/2 - Co_1_atom/1 -29.52
5.00 + dimer_sep_7/2 - Co_1_atom/1 -28.89
5.00 + dimer_sep_8/2 - Co_1_atom/1 -27.27
5.00 + dimer_sep_9/2 - Co_1_atom/1 -25.41
5.00 + dimer_sep_10/2 - Co_1_atom/1 -23.42
5.00 + dimer_sep_11/2 - Co_1_atom/1 -22.17
Not surprisingly the dimer entries of the bond stretch are single point calculations. With the default runtype being set to geometry optimization, single points can be requested by setting RUTYPE MAXIT 0 in the geo file:
BIOGRF 200
DESCRP dimer_sep_3
REMARK Dimer separation point #3
RUTYPE MAXIT 0
FORMAT ATOM (a6,1x,i5,1x,a5,1x,a3,1x,a1,1x,a5,3f10.5,1x,a5,i3,i2,1x,f8.5)
HETATM 1 Co 40.55000 40.55000 40.55000 Co 0 0 0.00000
HETATM 2 Co 39.45000 39.45000 39.45000 Co 0 0 0.00000
FORMAT CONECT (a6,12i6)
CONECT 1 2
CONECT 2 1
UNIT ENERGY kcal
ENERGY 3.445570
END
Description of crystaline phases¶
The training set includes equation of state (EOS) curves for the following crystaline phases
- hcp
- fcc
- bcc
- sc
- diamond
The curves were generated by performing a complete relaxation with a fixed cell volume. Within the file trainset.in the according energies are defined per-atom and set relative to the structure with the lowest energy. For example, the EOS for the hcp and diamond phases are defined as follows:
hcp phase

hcp entry in trainset.in:
# Volume Co_hcp
0.40 + EOS_hcp_6/2 - EOS_hcp_1/2 -13.42
0.20 + EOS_hcp_6/2 - EOS_hcp_2/2 -1.86
0.10 + EOS_hcp_6/2 - EOS_hcp_3/2 -1.04
0.10 + EOS_hcp_6/2 - EOS_hcp_4/2 -0.46
0.10 + EOS_hcp_6/2 - EOS_hcp_5/2 -0.12
0.10 + EOS_hcp_6/2 - EOS_hcp_6/2 -0.01
0.10 + EOS_hcp_6/2 - EOS_hcp_7/2 -0.08
0.10 + EOS_hcp_6/2 - EOS_hcp_8/2 -0.34
0.10 + EOS_hcp_6/2 - EOS_hcp_9/2 -0.77
0.10 + EOS_hcp_6/2 - EOS_hcp_10/2 -1.34
0.20 + EOS_hcp_6/2 - EOS_hcp_11/2 -2.00
0.40 + EOS_hcp_6/2 - EOS_hcp_12/2 -6.79
diamond phase
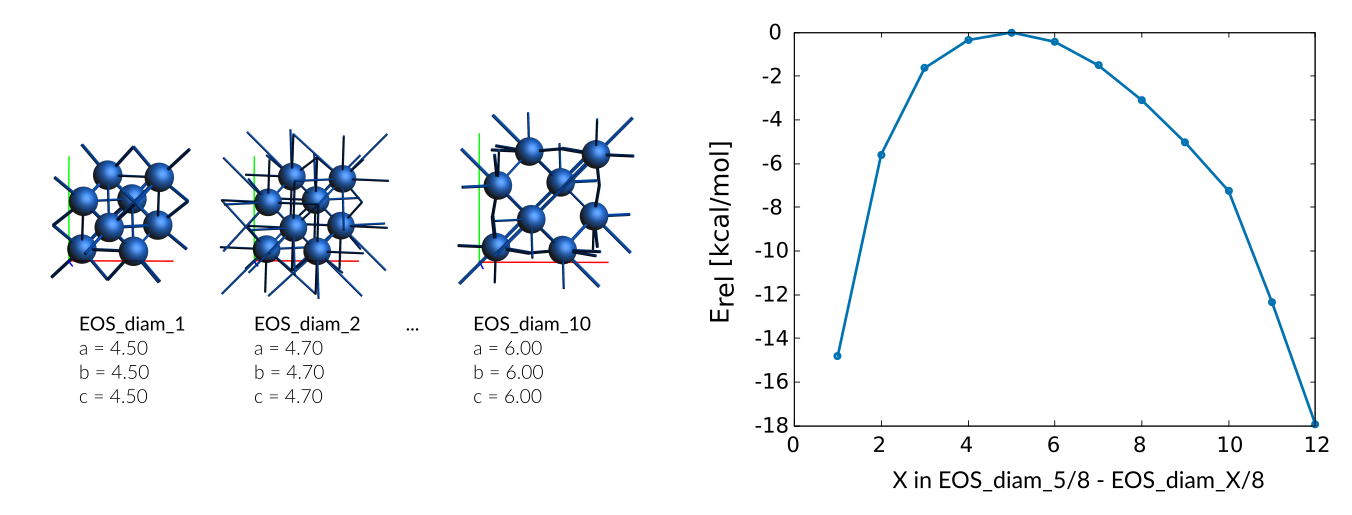
diamond entry in trainset.in:
# Volume Co_diamond
8.00 + EOS_diam_5/8 - EOS_diam_1/8 -14.80
8.00 + EOS_diam_5/8 - EOS_diam_2/8 -5.59
8.00 + EOS_diam_5/8 - EOS_diam_3/8 -1.63
8.00 + EOS_diam_5/8 - EOS_diam_4/8 -0.35
8.00 + EOS_diam_5/8 - EOS_diam_5/8 -0.01
8.00 + EOS_diam_5/8 - EOS_diam_6/8 -0.43
8.00 + EOS_diam_5/8 - EOS_diam_7/8 -1.51
8.00 + EOS_diam_5/8 - EOS_diam_8/8 -3.10
8.00 + EOS_diam_5/8 - EOS_diam_9/8 -5.04
8.00 + EOS_diam_5/8 - EOS_diam_10/8 -7.26
8.00 + EOS_diam_5/8 - EOS_diam_11/8 -12.35
8.00 + EOS_diam_5/8 - EOS_diam_12/8 -17.95
Description of Co-surfaces¶
The trainingset contains several surfaces which training value is a modified surface formation energy defined as the per atom energy of the surface relative to the energy per atom of the bulk phase (optimized hcp).
For the cubic surfaces (fcc,bcc,sc) both low-Miller and high-Miller surfaces are included. The (0001) surface has been added for the hcp phase only.
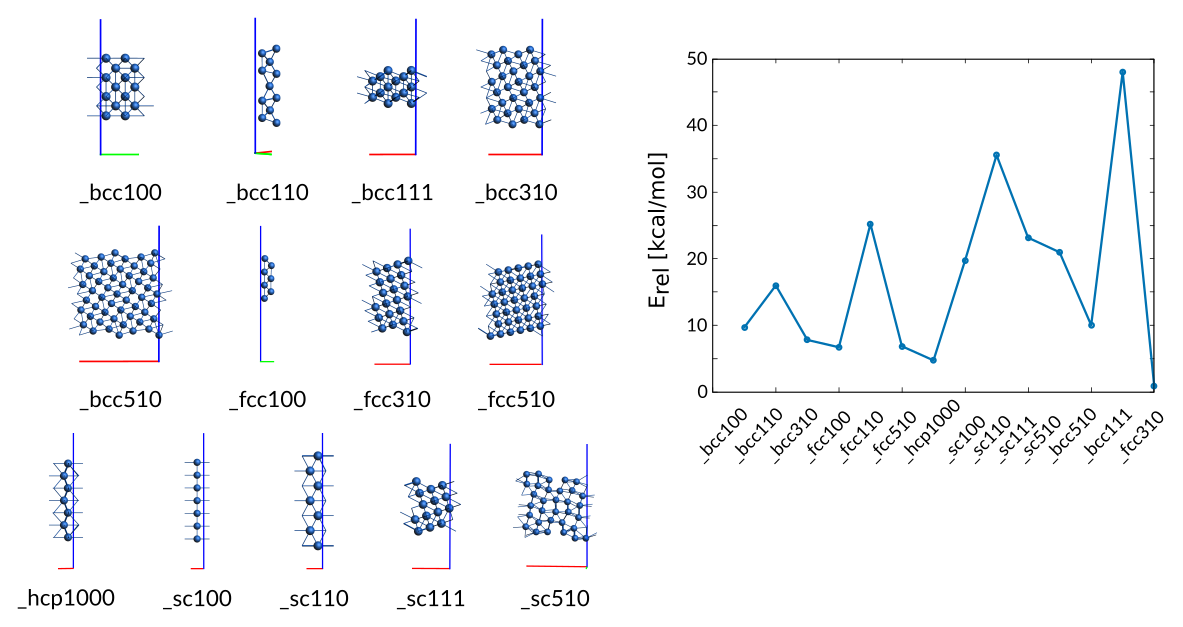
General Co surfaces entry in trainset.in:
# General Co_surfaces
2.00 + Surface_bcc100/28 - hcp_opt/2 9.67
2.00 + Surface_bcc110/11 - hcp_opt/2 15.97
2.00 + Surface_bcc310/30 - hcp_opt/2 7.79
0.10 + Surface_fcc100/14 - hcp_opt/2 6.69
8.00 + Surface_fcc110/7 - hcp_opt/2 25.20
2.00 + Surface_fcc510/37 - hcp_opt/2 6.84
0.05 + Surface_hcp1000/7 - hcp_opt/2 4.71
18.00 + Surface_sc100/7 - hcp_opt/2 19.72
18.00 + Surface_sc110/7 - hcp_opt/2 35.55
18.00 + Surface_sc111/14 - hcp_opt/2 23.14
18.00 + Surface_sc510/32 - hcp_opt/2 20.95
80.00 + Surface_bcc510/54 - hcp_opt/2 10.00
25.00 + Surface_bcc111/14 - hcp_opt/2 48.06
8.00 + Surface_fcc310/21 - hcp_opt/2 0.81
Tip
Creating various surfaces and bulk materials is easy with the GUI. See GUI tutorial: Building Crystals and Slabs .
Adatoms¶
Since the migration of Cobalt atoms on various surfaces is essential for the forming of energetically favorable surfaces, the trainingset contains a variety of adatom structures (top, bridge, hollow sites) on a variety of surfaces (fcc, bcc, sc). All DFT references were optimized, to ensure that the adatom is located in a local minimum.
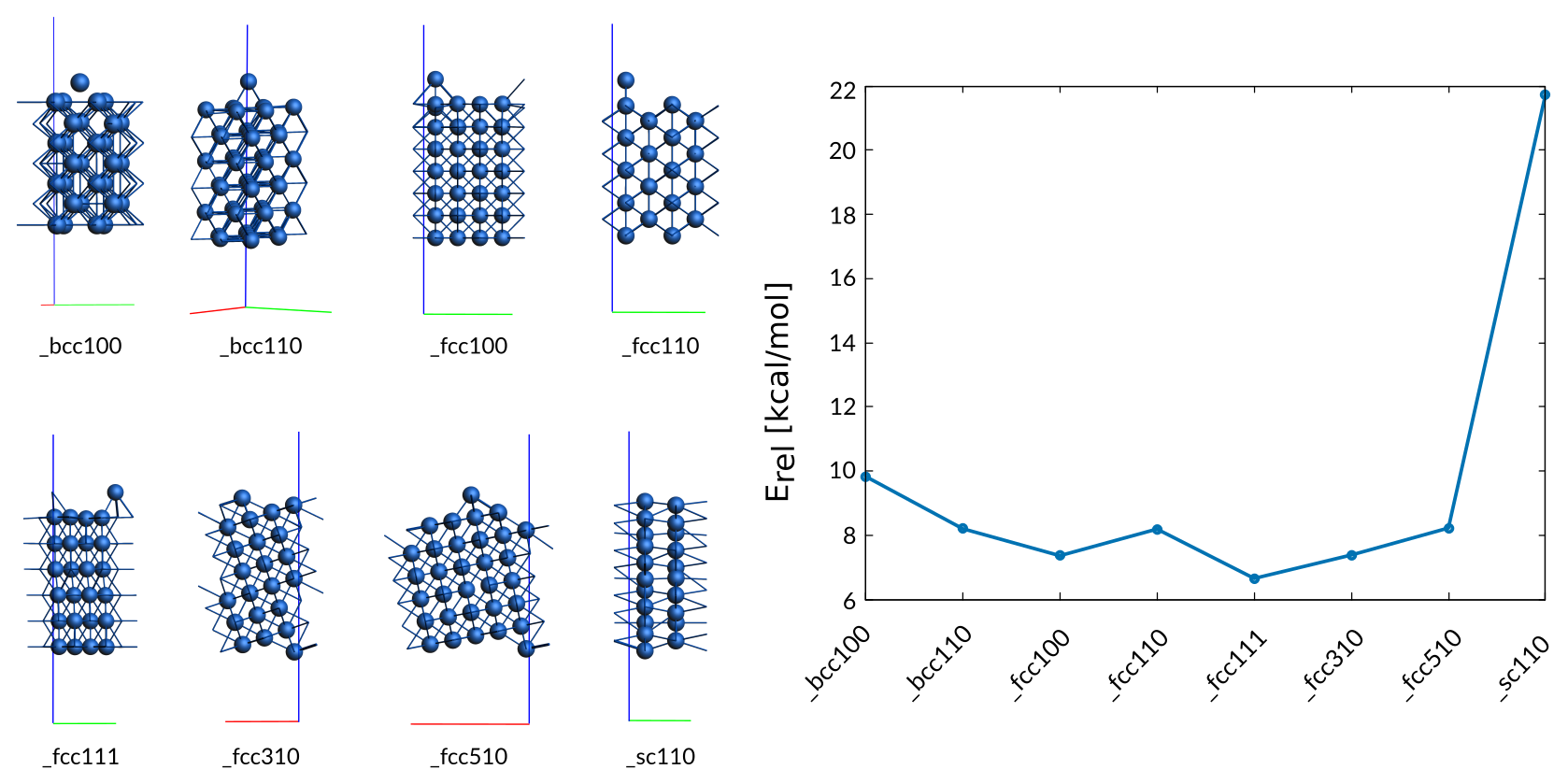
Co adatoms surfaces entry in trainset.in:
# General Co_surfaces_adatom
2.00 + Surf_adatom_bcc100/29 - hcp_opt/2 9.83
2.00 + Surf_adatom_bcc110/49 - hcp_opt/2 8.20
2.00 + Surf_adatom_fcc100/57 - hcp_opt/2 7.36
2.00 + Surf_adatom_fcc110/37 - hcp_opt/2 8.19
2.00 + Surf_adatom_fcc111/25 - hcp_opt/2 6.65
2.00 + Surf_adatom_fcc310/43 - hcp_opt/2 7.38
2.00 + Surf_adatom_fcc510/63 - hcp_opt/2 8.22
5.00 + Surf_adatom_sc110/29 - hcp_opt/2 21.74
Vacancies and defects¶
To consider vacancies and defects in bulk cobalt the training set contains
- bulk fcc cobalt with missing Co atoms (vacancies)
- amorphous bulk Co structures
- stacking fault defects
The trainingset contains formation energies of 1−6 coalesced vacancies in fcc cobalt. As discussed in the paper, the training data shows that it is most energetically favorable to have two vacancies as nearest neighbors.
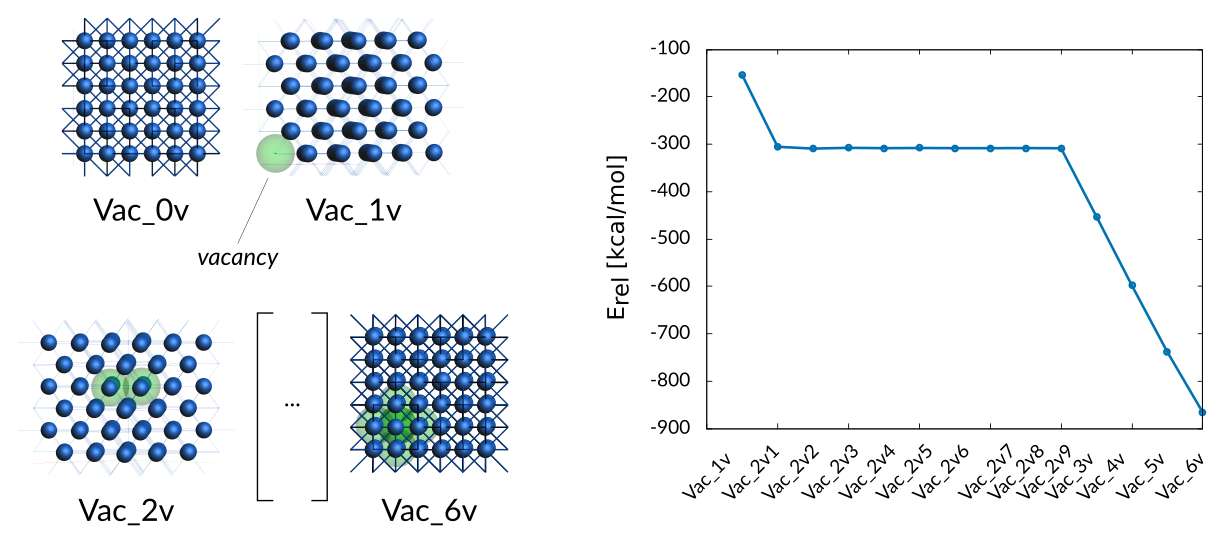
Co vacancies entry in trainset.in:
# General Co_vacancies2
1.00 + Vac_0v/1 - Vac_1v/1 -154.09
4.00 + Vac_0v/1 - Vac_2v1/1 -305.26
4.00 + Vac_0v/1 - Vac_2v2/1 -309.22
4.00 + Vac_0v/1 - Vac_2v3/1 -307.46
4.00 + Vac_0v/1 - Vac_2v4/1 -308.34
4.00 + Vac_0v/1 - Vac_2v5/1 -307.88
4.00 + Vac_0v/1 - Vac_2v6/1 -308.27
4.00 + Vac_0v/1 - Vac_2v7/1 -308.33
4.00 + Vac_0v/1 - Vac_2v8/1 -308.00
4.00 + Vac_0v/1 - Vac_2v9/1 -308.37
4.00 + Vac_0v/1 - Vac_3v/1 -452.51
4.00 + Vac_0v/1 - Vac_4v/1 -598.22
4.00 + Vac_0v/1 - Vac_5v/1 -738.33
4.00 + Vac_0v/1 - Vac_6v/1 -865.98
Amorphous bulk Co was generated by running ab-initio NVT MD at 2500K of 108 atom fcc lattice. The snapshots from the trajectory are included as single point calculations in the training data.

Amorphous entries in trainset.in:
# General Co_amorphous
8.00 + Amorphous_1/108 - hcp_opt/2 14.13
8.00 + Amorphous_2/108 - hcp_opt/2 10.35
8.00 + Amorphous_3/108 - hcp_opt/2 8.48
8.00 + Amorphous_4/108 - hcp_opt/2 7.27
4.00 + Amorphous_5/108 - hcp_opt/2 8.50
Stacking fault energies for the cubic phases were created via a half-lattice offset in [100] direction. The stacking fault energy for the hcp phase is described via a layering transition from hcp(0001) to fcc(111).

Stacking faults entry in trainset.in:
# General Co_stacking_faults
2.00 + SFE_bcc001/10 - hcp_opt/2 32.72
0.50 + SFE_fcc111/16 - hcp_opt/2 0.32
0.50 + SFE_hcp100/16 - hcp_opt/2 4.94
2.00 + SFE_sc001/8 - hcp_opt/2 21.28
Elastic strain moduli¶
As discussed in the paper elastic strain moduli are calculated by manipulating the lattice vectors describing the positions of the atoms. The following elastic constants c11, c12, and c44 for bulk Co phases are included in the training data:

Elastic moduli entry in trainset.in:
# General Co_elastic_constants
0.25 + Elast_bcc_c11/1 - hcp_opt/2 3.34
0.25 + Elast_bcc_c44/1 - hcp_opt/2 3.06
5.00 + Elast_diam_c11/2 - hcp_opt/2 29.89
5.00 + Elast_diam_c44/2 - hcp_opt/2 29.37
0.25 + Elast_fcc_c11/1 - hcp_opt/2 2.27
0.25 + Elast_fcc_c44/1 - hcp_opt/2 1.56
0.25 + Elast_hcp_c11/2 - hcp_opt/2 2.08
0.25 + Elast_hcp_c44/2 - hcp_opt/2 0.78
2.00 + Elast_sc_c11/1 - hcp_opt/2 18.74
2.00 + Elast_sc_c44/1 - hcp_opt/2 16.83
Tip
When manipulating the lattice in the graphical user interface (Model → Lattice) with the aim of inducing a strain, make sure you check the box Adjust atoms when changing lattice vectors. For the simulation of mechanical properties see also the advanced ReaxFF tutorial: Mechanical properties of an epoxy polymer .