DFTB charges, frequencies and dynamics (MD)¶
This tutorial will walk you through a few features of the DFTB engine, using the GUI. DFTB is computationally much faster than DFT, making it suitable for molecular dynamics evaluations, even on large systems.
We will walk through three different steps in this tutorial. The first step aims at pre-optimizing a simple molecule with DFTB. The second step will evaluate IR frequencies, spectrum and vibrational modes. In the third and last step we will perform a simple molecular dynamics calculation.
Step 1: DFTB: Pre-optimization and Charges¶
- Start ADFjobsSCM → New InputSelect the DFTB panel: panel bar ADF → DFTB
- In the ADFinput panel:cmd/ctrl-F, find benzeneSelect the entry Benzene(ADF) in the Molecule section of the search resultSelect the Main panelSet the model to DFTBSet the parameter directory to “Dresden” (normally you would want to use better parameters like the included 3ob set)Move your mouse over the parameter set option, and note the references in the help balloonClick ‘Pre-optimize’ in the DFTB panel to optimize the moleculeWhen done (DFTB Ready in the lower left corner of the window) use the View → Atom Info → Atomic charge: Net (dftb) → Show menu command to show the DFTB charges on each atom
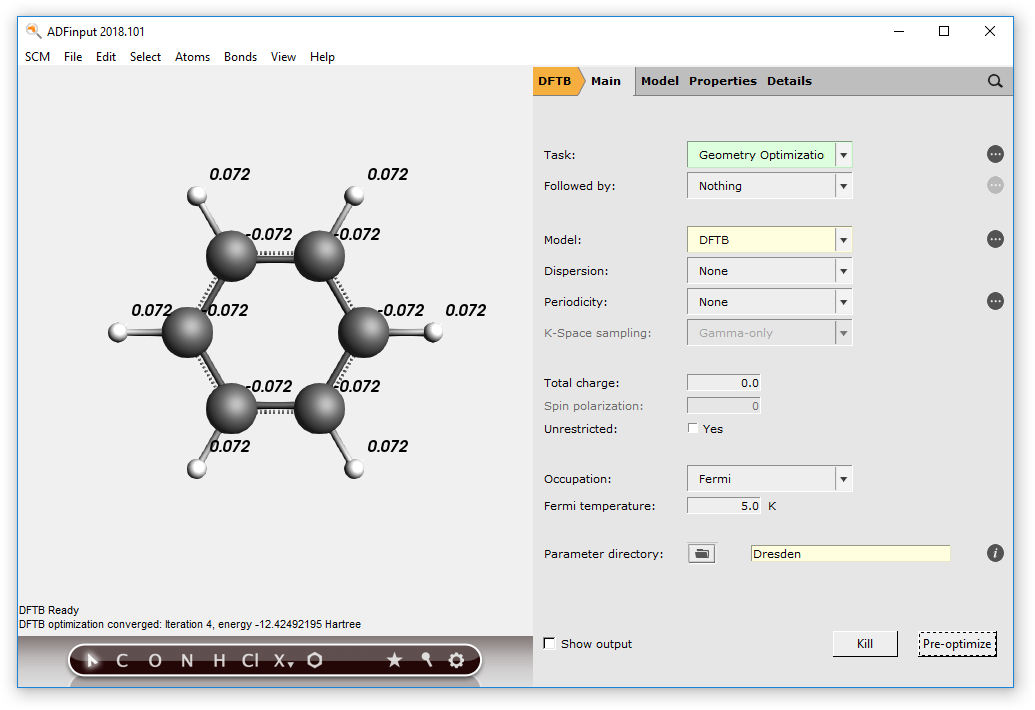
A Self Consistent Charge (SCC) evaluation allows atomic Mulliken charges to vary in an iterative procedure. This updates the DFTB Hamiltonian until self consistence of these charges is reached.
Enabling SCC requires higher computational cost due to the iterative procedure taking place for each energy evaluation, but leads to higher accuracy of the final result. If SCC is disabled, the resulting final charges are not self consistent, thus the procedure will be faster, but less accurate.
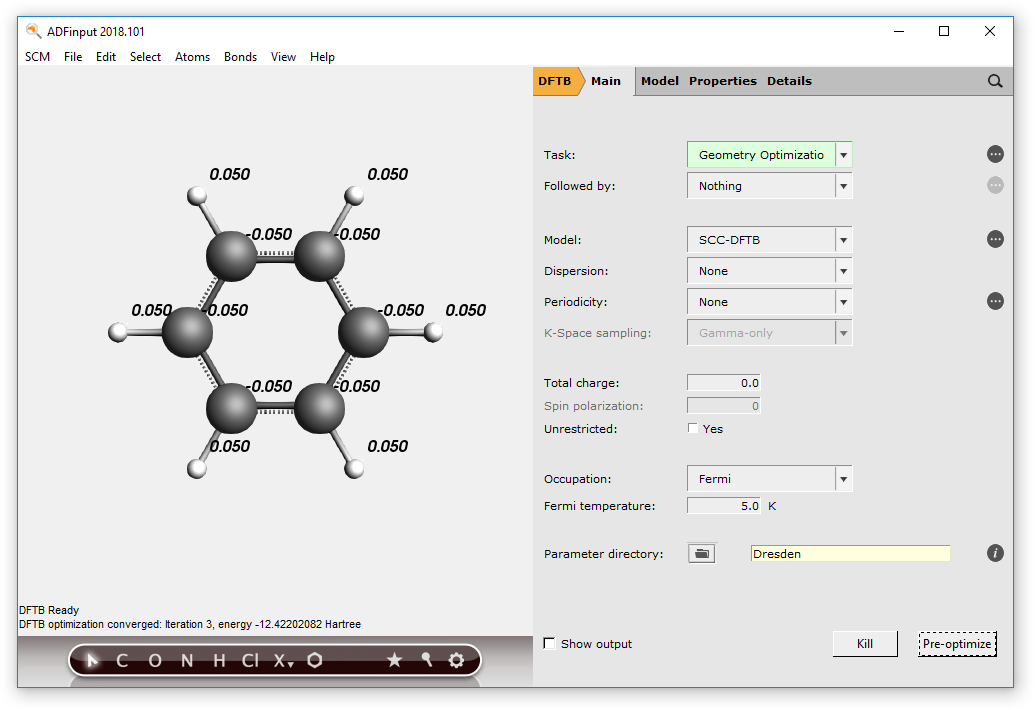
- Set the model to SCC-DFTBClick ‘Pre-optimize’ again in the DFTB panel to optimize the molecule.
Note how both equilibrium geometry and atomic charges are affected by the SCC evaluation:
Step 2: Frequency evaluation¶
This example will show how to compute vibrational frequencies and modes using DFTB.
- Choose Geometry Optimization as the Task to performChoose Followed By → Frequencies
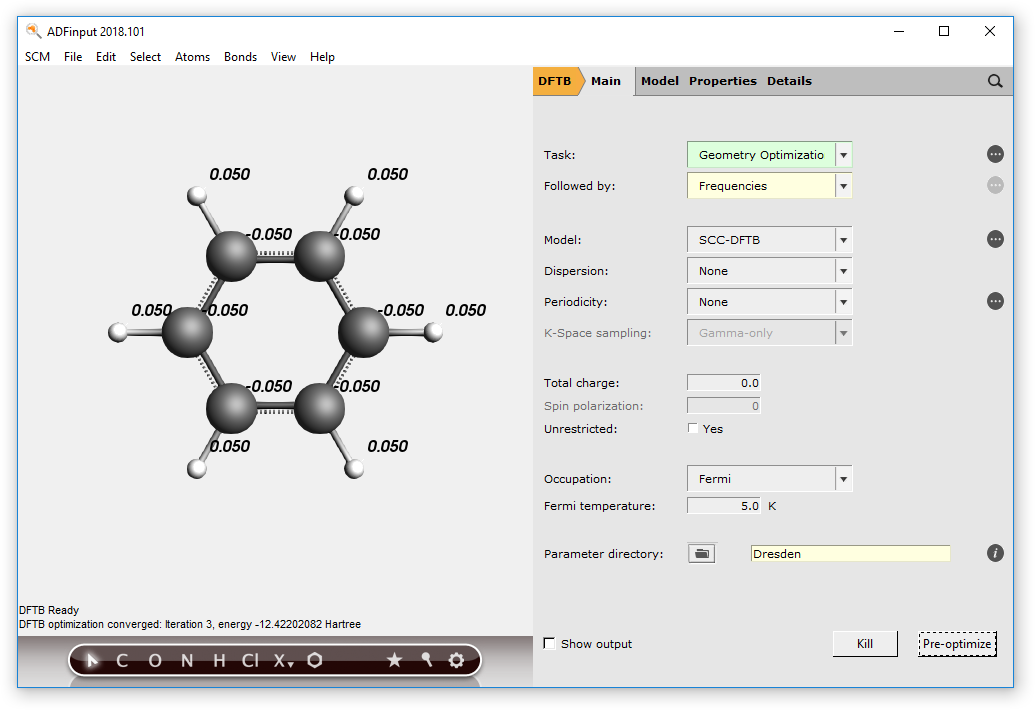
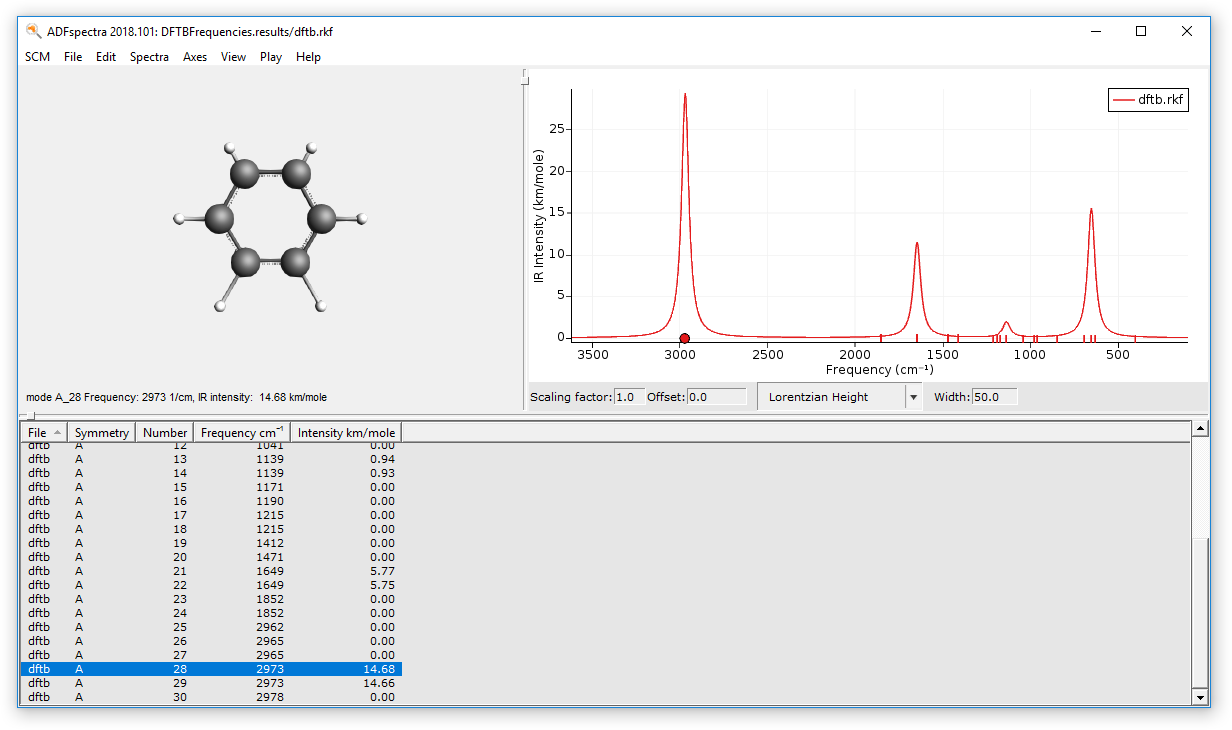
- File → Run, use DFTBFrequencies as name when asked for a nameIn ADFjobs:Select the “DFTBFrequencies” jobClick on SCM → SpectraClick on each individual frequency band to show the associated vibrational mode.
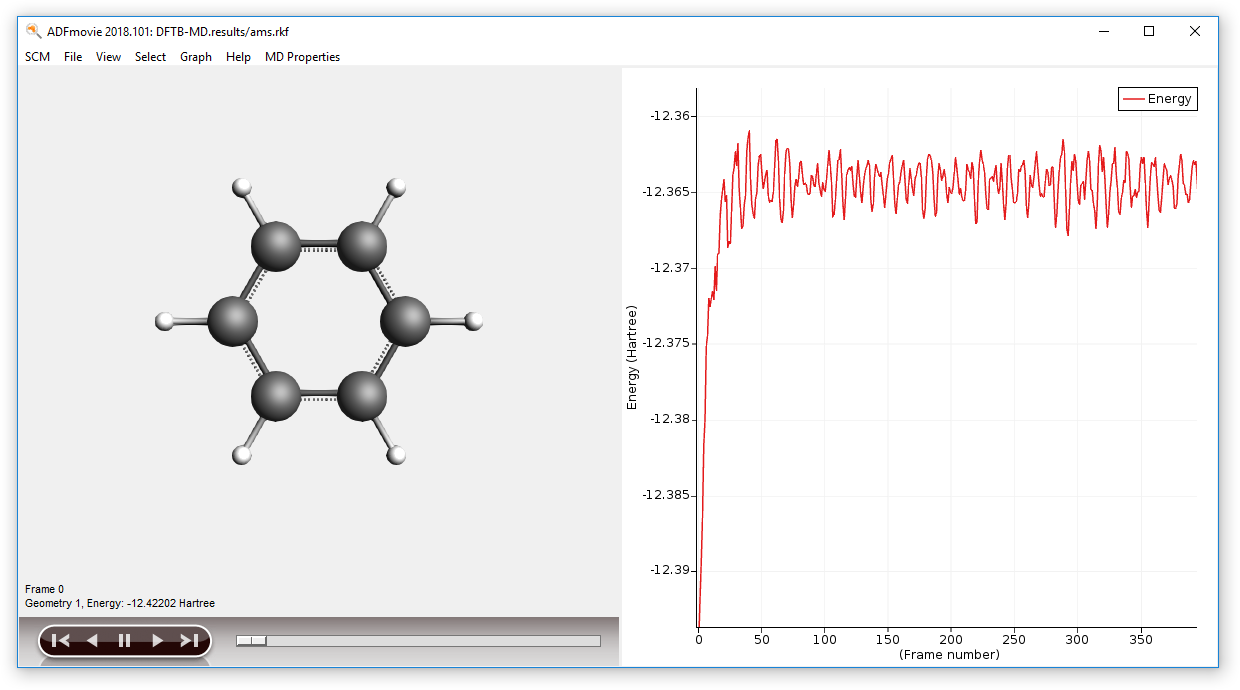
Step 3: Molecular dynamics¶
In this example, we will perform a simple molecular dynamics calculation using DFTB.
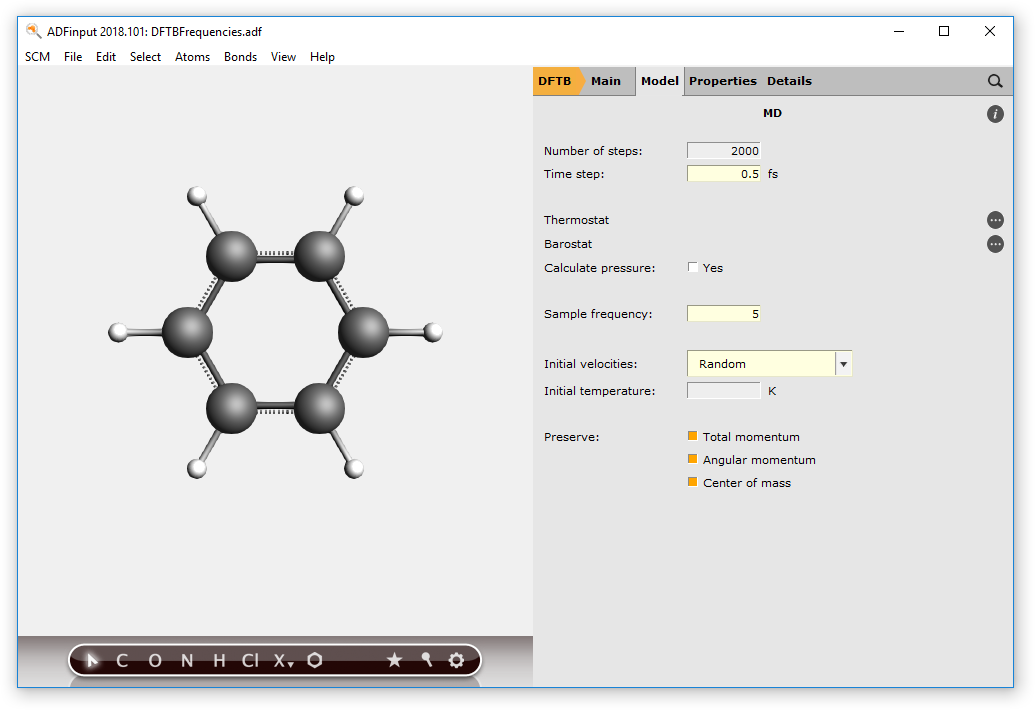
- In the ADFinput window, choose MolecularDynamics as the Task to perform.Choose Followed By → NothingMake sure the model SCC-DFTB option is selected, and the parameter set is Dresden (normally you want to use better parameters like the included 3OB set)Click on the Details button
 to the right of the Molecular Dynamics taskSet the Number of steps to 2000Set the Time step to 0.5set the Sampling frequency to 5Choose Initial velocities → Random
to the right of the Molecular Dynamics taskSet the Number of steps to 2000Set the Time step to 0.5set the Sampling frequency to 5Choose Initial velocities → Random
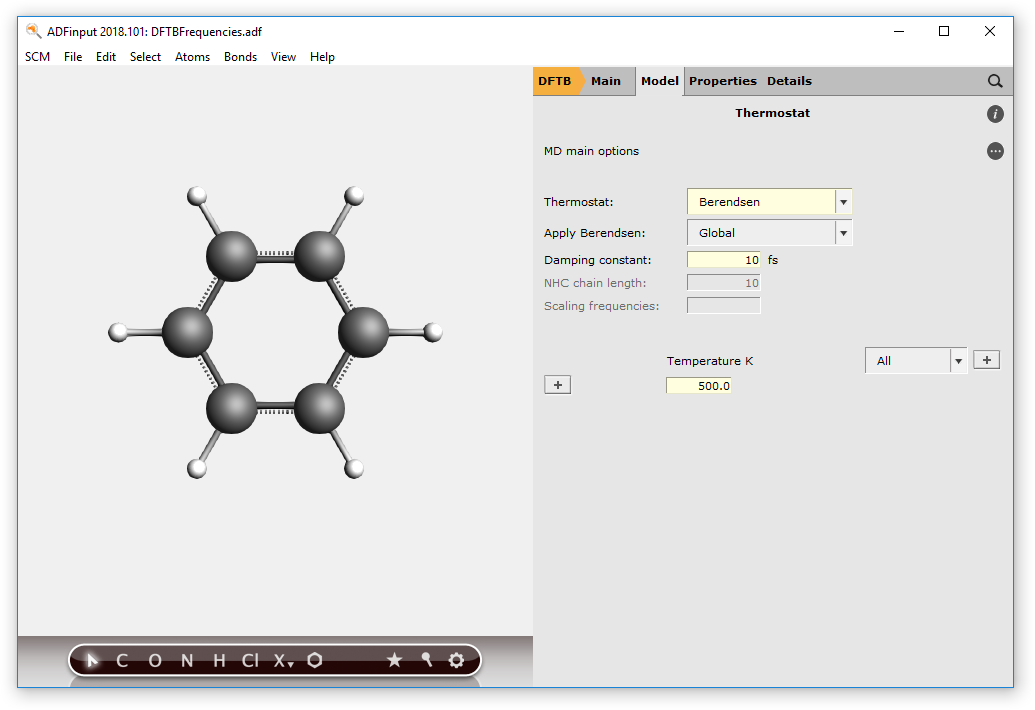
We now need to set the Thermostat options:
- Click on the Details button to the right ThermostatSelect Thermostat → BerendsenSet the Damping constant to 10 fsSet the Temperature to 500.0 kelvinSave your input choosing File → Save as ..., with name “DFTB-MD”
- File → run
You can monitor the calculation in real-time:
- While the calculation is running: SCM → Movie