Polymers¶
The COSMO-RS program can also be used to calculate thermodynamic properties of polymers and polymer-containing mixtures. The theory and equations behind this are described in the COSMO-RS documentation. This tutorial will focus on practical details of performing calculations with polymers in the COSMO-RS GUI.
This tutorial expects that that you are familiar with setting COSMO-RS parameters and pure compound data (see the COSMO-RS overview tutorial: parameters and analysis), doing COSMO-RS calculations (see the COSMO-RS overview tutorial: properties), and that the COSMO-RS compound database ADFCRS-2018 has been installed already (see the COSMO-RS compound database tutorial).
The ADFCRS Polymer Database¶
The COSMO-RS Polymer Database contains >200 structures of common polymers, all represented as the central unit of a trimer as described in the COSMO-RS manual. Because the simplest procedure used to generate the structure of a monomer still requires the calculation of a trimer, it is recommended to use the polymer database for this tutorial as adding user-defined polymers will likely be time-consuming (minutes-days, depending on the size of the polymer).
To quickly get started with polymers in COSMO-RS, download the COSMO-RS Polymer Database. More information about the database can be found here.
Selecting/inputting database compounds¶
After downloading and unzipping the database, open the COSMO-RS GUI AMScrs. To use the database in COSMO-RS, follow the steps shown below.
- Open a new COSMO-RS GUI windowSelect Compounds → Add Compound(s)Navigate to the Polymer Database directoryClick on monomers.compoundlist and select Open
Note
Polymeric compounds can also be added by SMILES string. Be sure to input a polymer in CurlySMILES format. One can read the CurlySMILES paper for more thorough coverage, but the CurlySMILES format is fairly self-explanatory. For example, in CurlySMILES polyethylene is C{-}C{n+} and polysytrene is C{-}C{n+}(c1ccccc1). These examples show that the atoms that are part of the polymerization are marked with {-} and {n+} identifiers.
Inputting your own polymers (optional)¶
It is also possible to input new polymers to the database and use these structures as you would any polymer in the database. As for all new compound inputs, there are two ways of doing this.
Method 1: Using ADF¶
This is the standard method for using compounds not already present in the database. This method requires an ADF and COSMO calculation, but polymer calculations can require a lot of atoms, sometimes making these calculations expensive. For this method, we must first perform a geometry optimization and then a single point COSMO calculation. The required steps for this calculation are as follows:
- Start AMSinput (SCM → New Input)Select Task → COSMO-RS CompoundInput the structure of a single monomer unit of your polymerAdd dummy (“Xx”) atoms corresponding to where the monomer is attached to other monomers in the polymeric structureGo to Model → Solvationclick “Generate” next to “COSKF trimer”Save/run your input file
We will illustrate the process of new polymer input using polyethylene below:
(1) Select Task → COSMO-RS Compound, Input the monomer structure
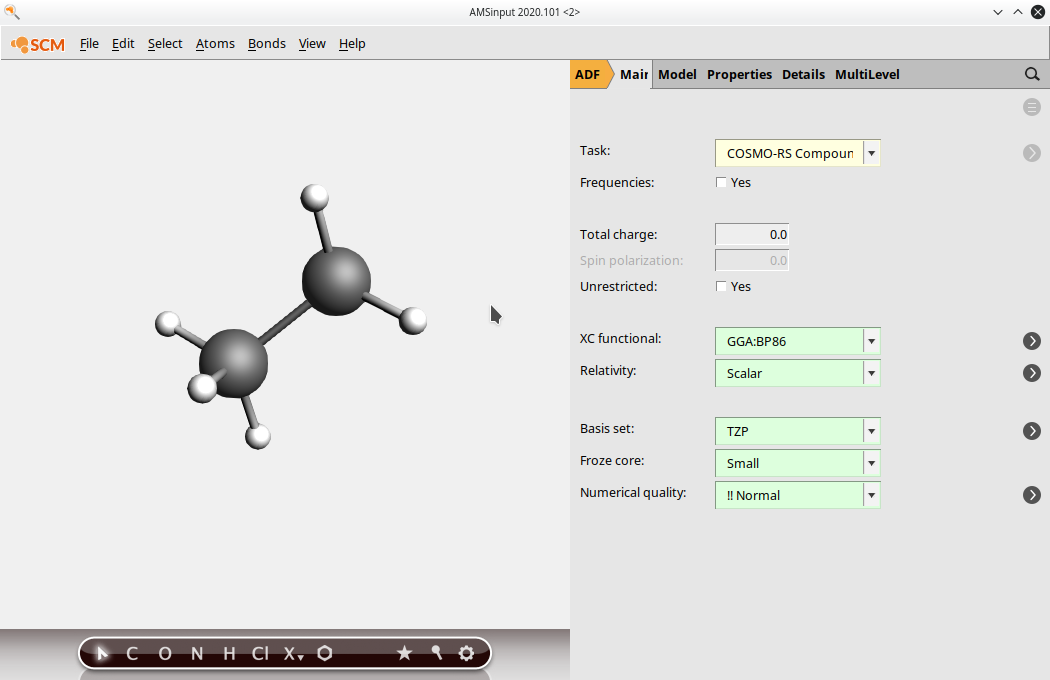
(2) Select the atoms that should be replaced by other monomer units in the full polymer. Note: depending on how you have inputted the structure, you may have to add these atoms. Be sure to check that all of the correct atoms are present. Select multiple atoms by holding the Shift key and clicking.

(3) Click on Atoms → Change Atom Type and replace the atoms with “Xx”

(4) Go to Model → Solvation and click “Generate” next to “COSKF trimer”
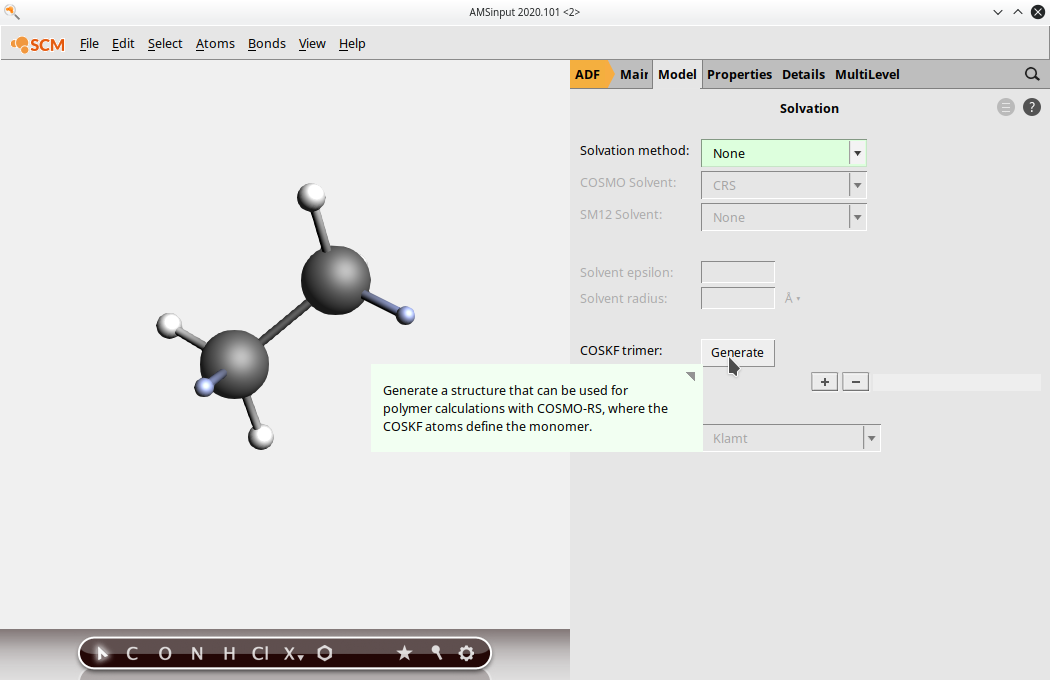
(4.5) (Optional, but recommended) Go to the DFTB menu, select GFN1-xTB in the Model field, and press the Pre-optimize button. This will provide a good initial geometry for the subsequent ADF calculation.

(5) Go back to the ADF panel and select Task: COSMO-RS Compound save your input file
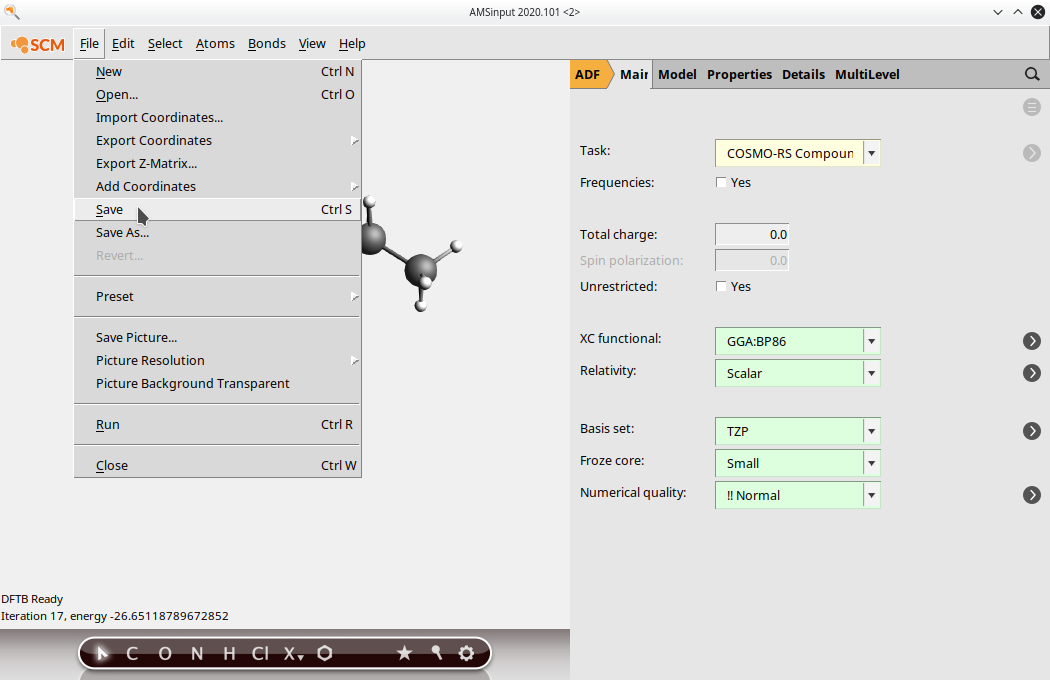
(6) Run the job (File → Run). After running the job, open the newly created .coskf file in the COSMO-RS GUI. Note that depending on the order of opening GUI windows the .coskf file may already have been opened by the COSMOS-RS GUI. You can see the COSMO surface by first selecting your new compound on the Compounds → List of added compounds window. Then press the “Show 3D” button, which opens an AMSview window. From AMSview, click Add → COSMO: Surface Charge Density → On COSMO surface points. This could produce the image below:

Method 2: Using FastSigma to estimate a polymer’s sigma profile¶
Because ADF calculations for large polymers can be expensive, it is sometimes preferred to estimate the necessary parameters to perform a COSMO-RS calculation. This can be done with the FastSigma tool available from the GUI or the command line. To use this tool from the COSMO-RS GUI, use the following steps:
- Select Compounds → List of added compoundsIn the top left corner in the SMILES input field, type the CurlySMILES for polystyrene
C{-}C{n+}(c1ccccc1)Click Add and save the file
The above steps should involve the following (main) step:

Inputting necessary property values¶
In the rest of the tutorial only polymers from the polymer database will be used.
Polymer/Average Molecular Weight¶
Because polymers of the same type can come in many lengths, it is important to be able to adjust the average molecular weight of a polymer. This is done from the compounds menu: Compounds → List of Added Compounds. Below, we show an example of setting the average molecular weight for Poly(ether_ether_sulfone) (PEES):

Additionally, to specify that a compound should be treated like a polymer, the Polymer box should be checked (shown in the image above). The number of repeat units will be set to the average molecular weight divided by the molar mass of the compound (the compound is assumed to the be monomer).
Density¶
Due to a modified combinatorial term for polymers, densities are required for every species in a calculation involving any polymers. In the ADFCRS database, many polymers already have a recommended density value, which should provide reasonable results under standard temperature and pressure conditions. However, it is well-known that for many polymers these density values can be sensitive to temperature and solvent. In these cases – or in cases where there is no recommended density value given – the density can be input from the compounds menu: Compounds → List of Added Compounds.
Here, we show an example of inputting the density for Poly(ether_ether_sulfone) (PEES):
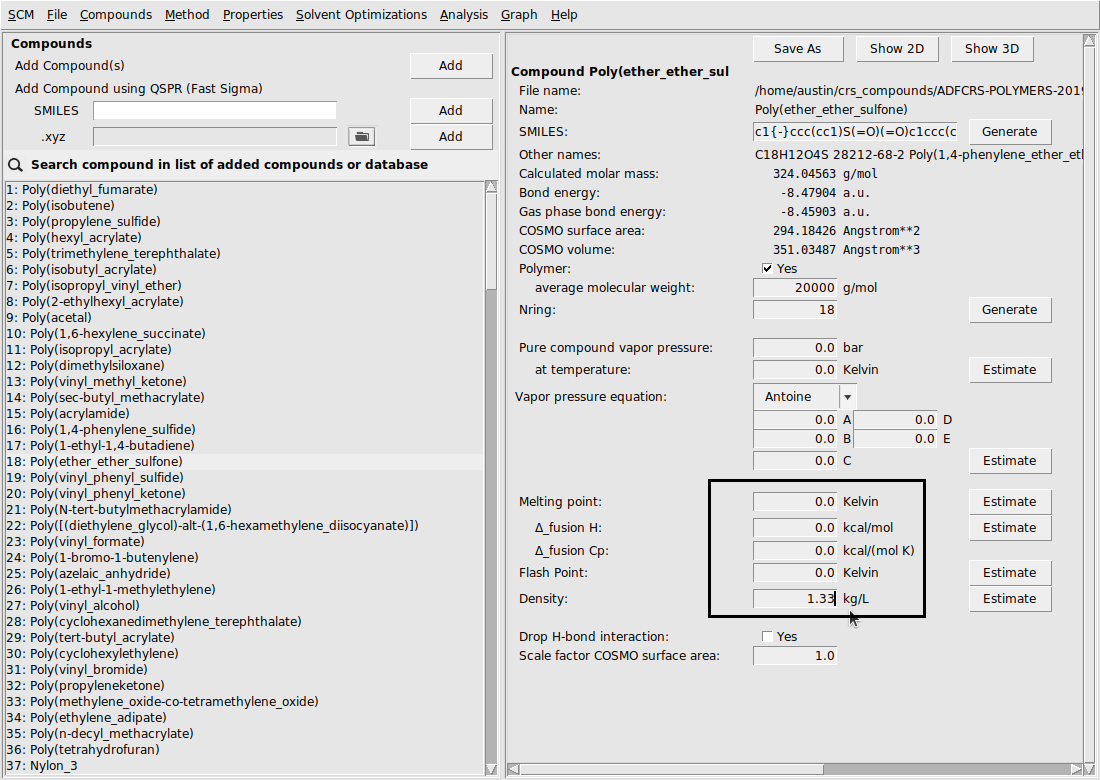
Note
Many polymers in the Polymer Database already have a given experimental density value. This value is automatically used for the calculation if the user does not input a different value for density. These experimental values come from the the Polymer Database Website.
Example polymer calculations¶
Polymers can be used just like other compounds in COSMO-RS/-SAC: they are simply selected as pure components or components of a mixture. If any polymers are present in the system (as indicated by the Polymer flag on the List of Added Compounds menu), then a number of special properties will be calculated. Because polymer calculations are done just like calculations for small molecules, this tutorial will cover a few example calculations without going into the details of every possible type of calculation. For a comprehensive tutorial about different problem types with the COSMO-RS GUI, we redirect the reader here.
Activity coefficients¶
For this calculation, we estimate the activity coefficients in a Poly(ethylene)/Benzene mixture. Because this is a calculation involving polymers, we will need the density of both Poly(ethylene) and Benzene. There is a recommended value of Poly(ethylene) given (0.852), so we can use that value. For Benzene, we’ll enter a value of 0.876. Use the following summary of steps to perform the calculation:
- Compounds → List of Added CompoundsSelect BenzeneSet Density to 0.876Properties → Activity CoefficientsSelect 2 ComponentsSelect Mass fractionSelect Poly(ethylene) and BenzeneSet mass fraction of Poly(ethylene) to 0.5Set mass fraction of Benzene to 0.5Click RunClick OK to the ‘Warning: no solutes found’
The result of this calculation is the following:

Notice that some non-standard properties are given: weight fraction activity coefficient (= mass fraction activity coefficient), volume fraction activity coefficient, and Flory-Huggins \(\chi\).
Vapor pressure of a mixture¶
Now, we will estimate the vapor pressure of a mixture of Poly(dimethylsiloxane), Methanol, and n-Hexane. Use the following summary of steps to perform the calculation:
- Compounds → List of Added CompoundsSelect MethanolSet Density to 0.792Click Estimate next to Vapor pressure equationSelect HexaneSet Density to 0.655Click Estimate next to Vapor pressure equationProperties → Vapor pressure mixtureSelect 3 ComponentsSelect Mass fractionSelect Poly(dimethylsiloxane), Methanol, and HexaneSet mass fraction of Poly(dimethylsiloxane) to 0.5Set mass fraction of Methanol to 0.25Set mass fraction of Hexane to 0.25Adjust the temperature range from 298.15 K to 398.15 KClick Run
The result of this calculation is the following:

Note that estimated properties are calculated with the Property estimation tools
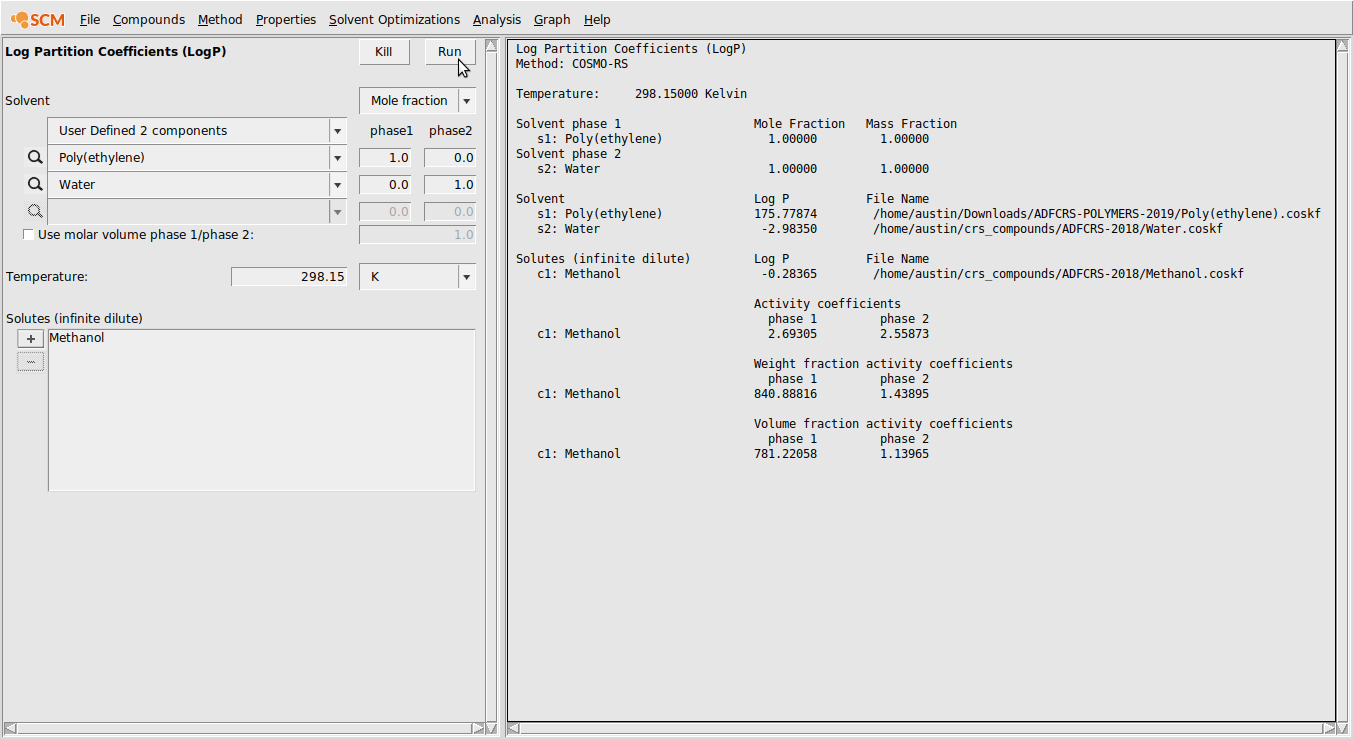
Partition Coefficients (LogP)¶
Now, we’ll calculate the partition coefficient of Methanol between a water and poly(ethylene) phase.
- Compounds → List of Added CompoundsSelect MethanolSet Density to 0.792Select WaterSet Density to 1.0Properties → Partition Coefficients (LogP)User Defined 2 componentsSelect Poly(ethylene) and WaterSet mole fraction Poly(ethylene) to 1.0 in phase 1Set mole fraction Water to 1.0 in phase 2Select Methanol in the Solutes (infinite dilute) boxClick Run
The output of this calculation is shown below:
Solubility in Pure Solvents¶
In this example, we’ll calculate the solubility of Hexane gas in Poly(styrene) over the temperature range 398.15 K to 498.15 K and over the pressure range 1.0 bar to 4.0 bar.
- Compounds → List of Added CompoundsSelect HexaneSet Density to 0.655Click Estimate next to Vapor pressure equationProperties → Solubility in Pure SolventsSelect Poly(styrene) in Pure Compound SolventsSelect Hexane as the SoluteSelect Gas for the solute typeSet the temperature range from 398.15 K to 498.15 KSet the number of steps for pressure to 3Set the pressure range from 1.0 bar to 4.0 barClick RunClick Graph → Y axes → Solubilty (g/L solvent)

Binary Mixture and Flory-Huggins \(\chi\)¶
In this example, we will calculate binary mixture properties (including the \(\chi\)) over the entire range of compositions. We will do this for a binary mixture of Benzene and Poly(ethyl_ethylene) and use COSMO-SAC (the 2013-ADF Xiong parameters).
- Compounds → List of Added CompoundsSelect BenzeneSet Density to 0.876Method → COSMO-SACMethod → ParametersSelect the 2013-ADF Xiong parametersProperties → Binary Mixture VLE/LLESelect Poly(ethyl_ethylene) as Compound 1Select Benzene as Compound 2Select Mass FractionClick RunClick Graph → X axes → w1: mass fraction 1Click Graph → Y axes → Flory-Huggins 𝛘
The result of this calculation is the following:
