Try out AMS2023 or upgrade your license?
If you already have an annual license for our software with an automatic license – just download a new binary. To update your cluster license, contact license@scm.com, mentioning your user ID. If you have an older perpetual license or would like to try out new modules, contact us for an upgrade quote or an evaluation license for 2023.
If you do not have any existing license, request a free trial.
Release notes of previous versions
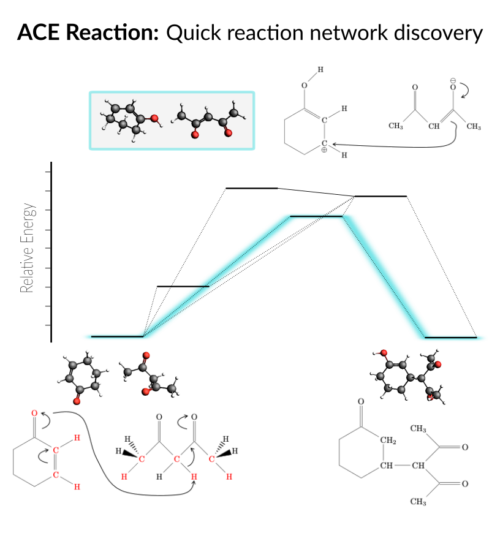
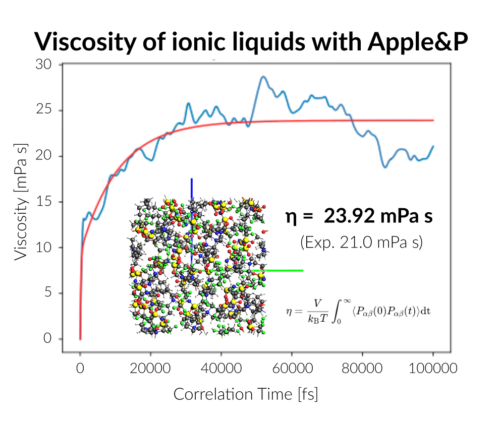
AMS2022: DFTB & ReaxFF parametrization, OLED workflows (deposition, properties), GFN-FF, qsGW, faster FCF, QM/FQFμ, reaction discovery, kMC interface, COSMO-RS-PDH
AMS2021: Automated reaction pathways, GW, polarizable force fields
AMS2020: Multi-layer (QM/MM), ML Potentials, G0W0, ADF-in-AMS, external electric fields for periodic systems
AMS2019.3: microkinetics, fast periodic DFTB, double hybrids, NEB, VASP interface
AMS2019.1: accelerated MD, Solvation Model 12 (SM12) for BAND, GFN-xTB, polymers with COSMO-RS
AMS2018: AMS driver for advanced PES tasks, new excitation capabilities in ADF, ReaxFF analysis and fitting
ADF Modeling Suite 2017: XCDFT, CV-DFT, X2C, periodic NEGF, 2D-TDCDFT, periodic DFTB3, DFTB fat bands, eReaxFF, NHC, MOF & COF builder, VCD analysis tools, molecule gun, Quantum ESPRESSO interface, COSMO-SAC-2016, iPy, improved ASE interface
ADF Modeling Suite 2016: CDFT, SM12, libXC interface, standard RSHs, XES with quadrupoles, TD-DFT+TB, sTDA, sTDDFT, MECP Polarization functional for semiconductor optical properties, periodic energy decomposition analysis, analytical lattice gradients, scripting with COSMO-RS, QuasiNano2015 DFTB parameters, TDDFTB gradients and FCF, ChemTrayzer, ACKS2
Request free trial