Electronic transport in a 1D gold chain¶
Introduction¶
The system studied in this tutorial is discussed in Electronic and Transport Properties of Artificial Gold Chains PhysRevLett.93.096404 and Benchmark density functional theory calculations for nano-scale conductance The Journal of Chemical Physics 128, 114714 (2016).
We will simulate the electronic transport through an atomic gold chain, and study the effect that an adsorbed CO molecule has on the conductance.
According to PhysRevLett.93.096404 “a single CO group […] modulates the electronic wave functions, acting as a ‘‘chemical scissor’’ along the gold chain, to strongly modify the coherent transport properties of the system”.
See also
Creating the lead file¶
Let’s begin by creating a lead file. A lead file is a simple .xyz file with an extra lattice vector.
Tip
The folder $AMSHOME/atomicdata/Molecules/NEGF/Leads contains some pre-defined lead files
For this tutorial the lead will be a single gold atom with a lattice of 2.9 Å (in the x-direction). Let us create this with AMSinput:
 →
→ 
 to switch the Lattice panel
to switch the Lattice panel
In the lattice panel we can specify the lattice vector:
We now add the gold atom
and set the coordinates of the gold atom to (0,0,0):
We now export this 1D gold chain as an .xyz file:
The .xyz file, defining our lead, should look like this:
1
Au 0.00000000 0.00000000 0.00000000
VEC1 2.90000000 0.00000000 0.00000000
Gold chain transport calculation¶
We are now ready to set up the NEGF calculation for the gold chain. The simulation in this tutorial can be performed with either the DFTB engine or the BAND engine. DFTB is computationally faster than BAND, but the results will generally be less accurate.
→  next to Task: NEGF to switch to the NEGF panel (or click on Model → NEGF) → next to Task: NEGF to switch to the NEGF panel (or click on Model → NEGF)
next to Task: NEGF to switch to the NEGF panel (or click on Model → NEGF) → next to Task: NEGF to switch to the NEGF panel (or click on Model → NEGF)In the NEGF panel, import the lead file we just created (‘Au_lead.xyz’):
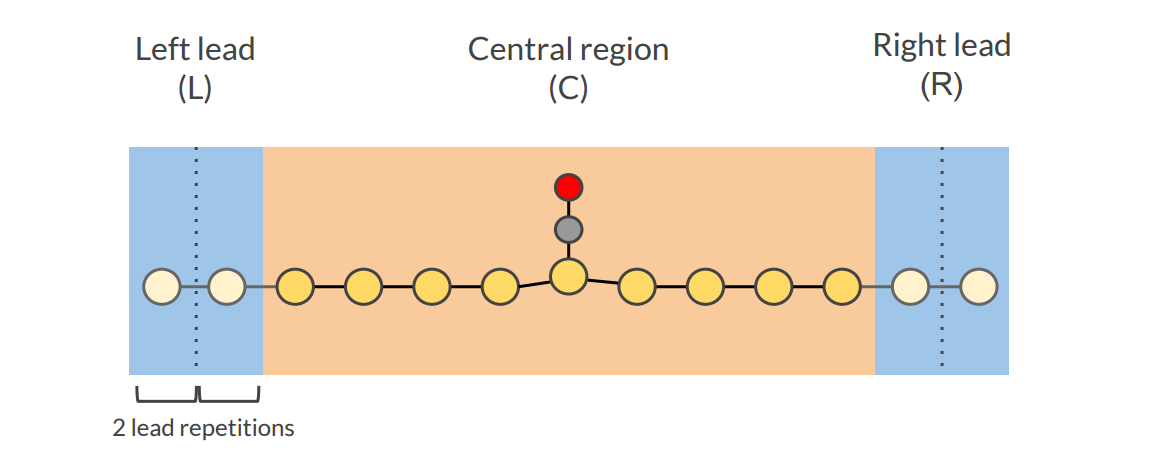
Fill the central region with 9 gold atoms using the “Fill central region” option:
Let us also change the range for the Transmission energy grid to [-3.5,3.0], to match the energy range of PhysRevLett.93.096404:
This is what your set-up should look like:

We are now ready to run the calculation and visualize the results with AMSspectra:
This is the computed transmission function through a 1D gold chain:
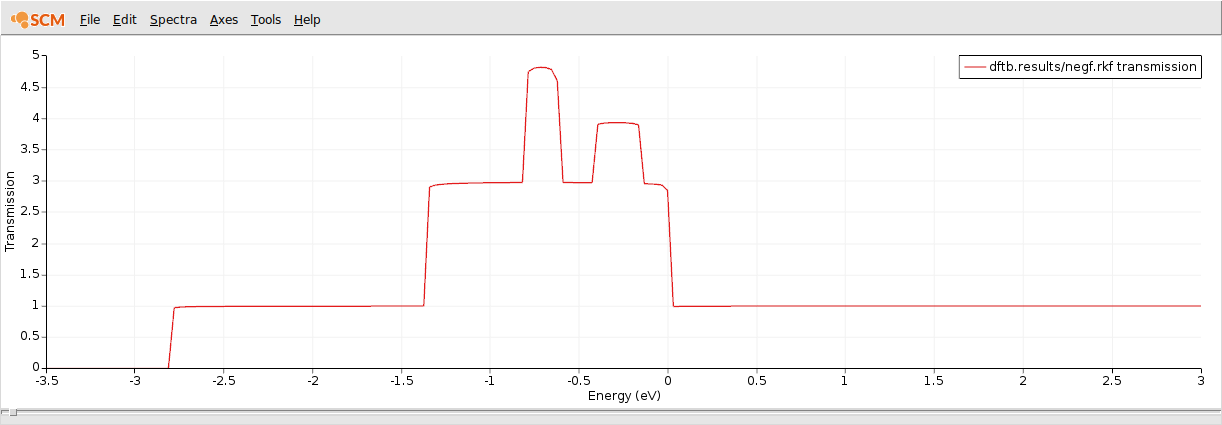
CO on gold chain transport calculation¶
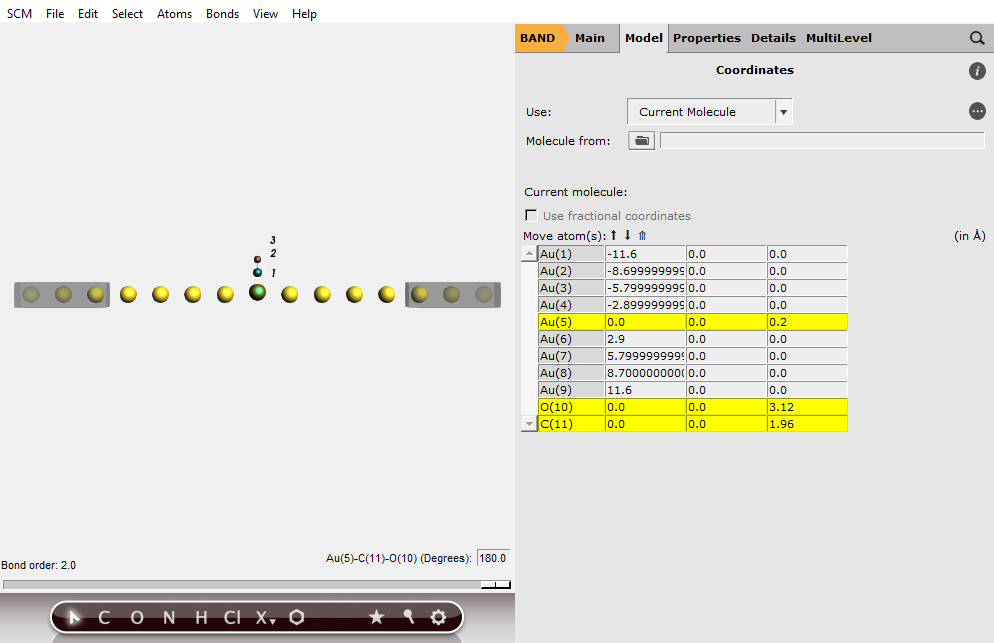
We now modify our previous system by adding a CO molecule in the central region:
O 0.0 0.0 3.12
C 0.0 0.0 1.96
The gold atom on which CO is adsorbed is “pulled” towards the CO molecule by 0.2 Angstrom:
Your system should look like this:
Tip
It is good practice to include some buffer lead material in the central region, and test the convergence of the results with respect to the buffer size (in this tutorial we have 4 buffer gold atoms on each side of the central Au-CO).
Run the calculation and visualize the results with AMSspectra:
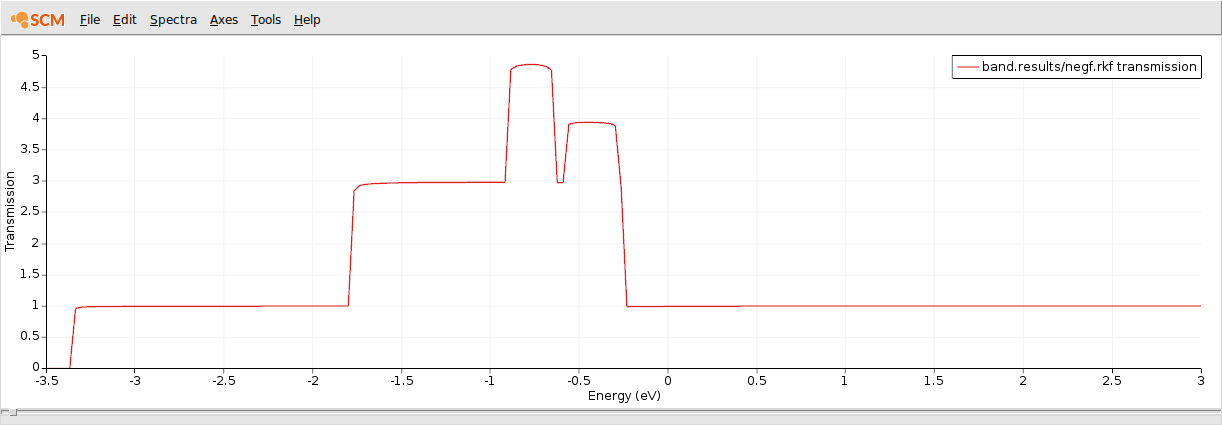
This is the computed transmission function when CO is adsorbed on a gold chain.
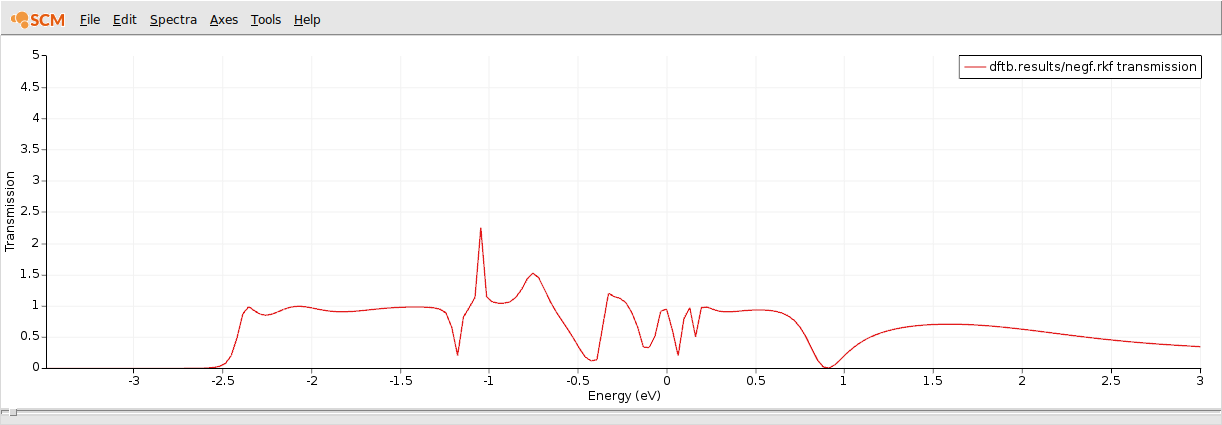
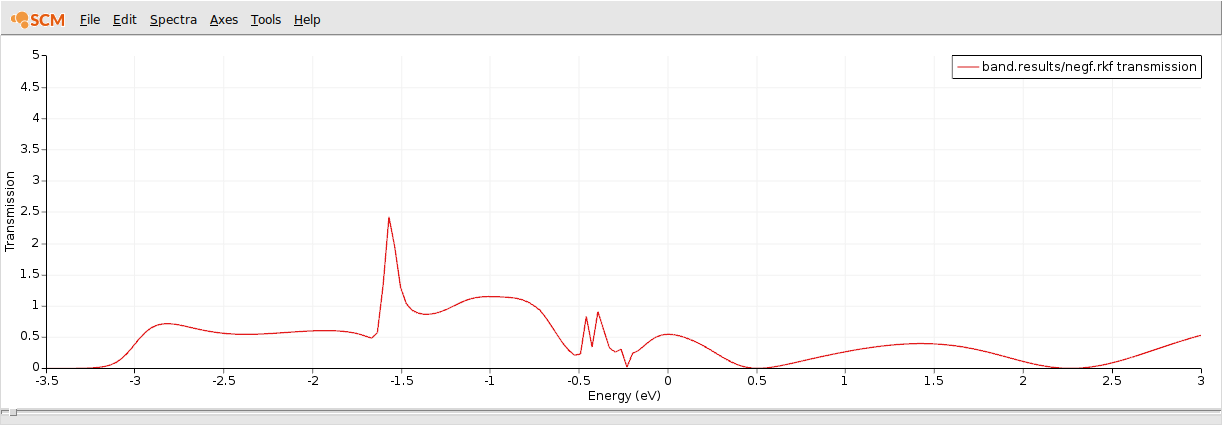
As expected, the conductivity around the Fermi energy is suppressed by the adsorbed CO molecule.